Credit: Riken Research
Next-gen sequencing technologies have enabled rapid identification of many genes contributing human disease. Rapid, inexpensive exome sequencing quickly gave us access to the low-hanging fruit: rare Mendelian disorders with single, highly penetrant coding mutations. Since 2009, we’ve seen an avalanche of reports of disease-causing mutations and novel disease genes. Family studies, case-control studies, and population cohorts are picking up this kind of signal everywhere.
The trouble, as anyone who’s analyzed this kind of data understands all too well, is that there are a lot of possibilities out there. You can take just about any gene from a sequencing study or GWAS and — with the assistance of a nice resource like Gene Cards — come up with a story that might connect mutations/variants in that gene to your phenotype of choice. But the burden of proof remains.
Functional Validation Required
It should now be obvious to most that publication of novel disease genes in top-tier journals requires more than just genetic or genomic data. It requires some kind of functional validation, an assay that demonstrates how genetic differences have a measurable phenotypic effect that makes sense for the disease. Genomic and statistical approaches are hypothesis generation tools. Those hypotheses, well-supported as they may be, must be tested in vitro or in vivo to see if they hold up. Because, as I said, you can spin a story about almost any gene.
Let’s say that you’ve identified a new possible cancer susceptibility gene, a candidate tumor-suppressor. You found it by looking for rare germline variants in a cohort of patients with a specific form of cancer. You’ve already done the genomics to establish that:
- Rare variants in the gene are enriched among cases (maybe 5% of patients harbored rare deleterious variants in that gene, compared to 0.1% of 1000 Genomes or NHLBI-ESP populations).
- In tumors, the gene is a target for biallelic inactivation by somatic mutation, deletion, LOH, or epigenetic silencing
- Expression of the gene is reduced or ablated in affected patients or tissues.
Everything looks right, it sure looks like a tumor suppressor, but where’s the proof? With over 20,000 known protein-coding genes, widespread genetic variation, and the continual accumulation of mutations in somatic tissues, there are plenty of candidates that will meet these criteria by chance alone. Editors and reviewers of top-tier journals know this, and they want more. They want functional tests demonstrating that defects in your gene improve the growth, survival, proliferation, or metastatic potential of cells. They want a null mouse for your gene that’s prone to tumors. As much as it pains me to say it, the following statement is true.
Genomics is not enough.
Options for Functional Validation
This bitter medicine undoubtedly tastes sweet to the molecular biologists and bench scientists whose efforts may have been overshadowed by genomics in recent years. Because now, after all of our fancy high-throughput instruments, robust informatics and clever statistics have provided some answers, we have to leave the computer and head back to the laboratory. And many of us, including the author, have little to no experience there.
Even so, I’ll do my best to summarize some of the options for functional validation, and ask you readers to comment with the things I’ve gotten wrong or forgotten.
Molecular Assays
Subcellular localization (Weiqiao, PNAS 1998)
Additional evidence can be garnered at the molecular level by showing that your gene functions
- mRNA expression. Genome wide (RNA-seq) or targeted (RT-PCR) mRNA expression assays provide insight about gene expression at the transcript level, including exon usage and alternative splicing.
- Transcript/protein localization. It has been possible for some time to examine the tissue and/or intracellular location of a protein using specific dye-tagged antibodies, which may lend support to the idea that your gene of interest plays an important role at that location.
- Protein-DNA interaction. New, high-throughput chromatin immunoprecipitation and sequencing (CHiP-Seq) make it possible to identify sequences bound by specific proteins. This can be used to evaluate the protein that does the binding (showing that a variant alters when/where/how it binds) or the target regulatory sequence (showing that variants affect binding of an important regulatory protein, such as a transcription factor).
- Protein-Protein interaction. Another intriguing possibility for functional validation is showing that your suspect gene encodes a protein that interacts with a known key player in your disease pathway, such as BRCA1/2 for homologous DNA repair in breast and ovarian cancers.
Biological Assays
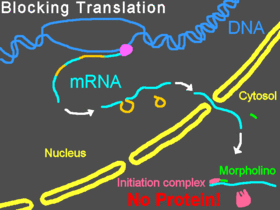
Morpholino Knockdown (Wikipedia)
Functional validation of a candidate disease gene can also be performed in living cells or organisms. Often this garners more compelling evidence of a gene’s importance, because it demonstrates the relationship between a genetic entity and phenotype visible at the cellular level or above. Some of the approaches here include:
- Human cell lines. Gene knockdown (by siRNA or other methods) or transfection (infection of cells with a virus genetically engineered to carry a certain gene) in cell lines serves to demonstrate its importance for measurable cellular phenotypes, such as apoptosis, growth, proliferation, and contact inhibition.
- Animal models. We are lucky enough to control the fates of lesser organisms, which means we can use reverse genetics techniques to alter their genomes and see what happens. The advantage here is that you get to study a gene’s effect on a complete organism, which more closely resembles what could be happening in humans. Mouse models are often the method of choice, though some other model organisms provide good experimental systems for certain phenotypes, such as morpholinos (antisense oligos) in zebrafish.
- Human patients. This generally isn’t possible, but in some cases genetic information (i.e. specific tumor alterations) has been used to tailor treatment to individual patients, in which case the outcome of the treatment validates the genomic finding. Case in point: the use of whole-genome sequencing to diagnose a cryptic PML-RARA fusion. This approach obviously has many ethical and legal hurdles, and probably wouldn’t be approved for truly novel discoveries.
A Call to Reviewers
In closing, I would like to appeal to peer reviewers of those journals who now wish to see functional validation of genomic findings. Asking the authors to “provide some functional validation” of their findings may be a valid critique, but it’s not terribly helpful. It would be better to outline what kind of experiments you’d like to see to become convinced. Because the odds are, you’ll be reading this manuscript again at some point, and wouldn’t it be nice if they performed the validation that you were looking for?
In fairness, some of those who work in the field of next-gen sequencing, even to tackle genetic diseases, do not have knowledge of (or even access to) laboratory techniques that could functionally validate their findings. It would benefit the entire research community if we took a moment to outline potential avenues of functional validation so that we “dry lab” scientists can begin to explore them.









