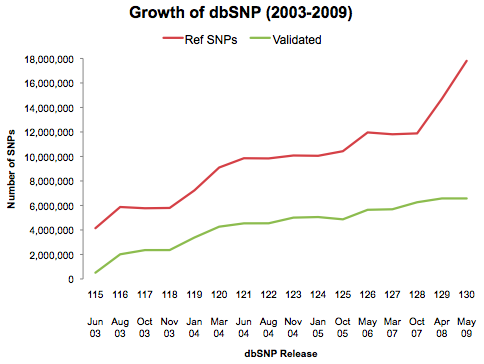
The rapid advance of next-generation sequencing technologies, particularly in the last several years, has almost seemed like something out of a science fiction novel. Think about it: on a HiSeq X Ten instrument, we can sequence a complete human genome in less than a week, at a cost that’s 0.00001% of what it took to fund the Human Genome Project.
It might surprise you to learn that — in addition to my blog posts here, and the grant/paper writing I do for my job — that I dabble in science fiction writing as well. If you think that scientific publication/success is hard (10% acceptance rate for tip-tier journals, or 8% NIH funding level), you should look into the the fiction side of publishing sometime.
The acceptance rate for most professional science fiction magazines (for short fiction) is generally below 1%. The pay is usually $0.05-$0.10 per word, meaning that a 4,000 word story might bring $200-400 in the (unlikely) event that you get it professionally published. The odds of landing a literary agent — which is required, if you want to have your novel shopped to most traditional publishing houses — are about 1 in 1,000.
A few months ago, Third Flatiron Publishing (which does quarterly science fiction anthologies) announced that their Spring 2015 anthology would be themed around world-altering events. As it happened, I’d written a science fiction story that seemed like it might fit — it was about a couple of researchers working in a dusty lab who stumble upon a universal cure for cancer (you remember I said science fiction, right?), and their struggle to make it available to the world.

The Time It Happened
I’m thrilled to say that the editors at Third Flatiron liked my story enough to choose it for their anthology The Time It Happened, which just came out and is available on Amazon in both Kindle and paperback versions. They’ve also bought audio rights, and intend to create a free podcast of my story (as well as a couple of others) sometime in the near future.
Since you readers enjoyed the non-fiction I write for MassGenomics, hopefully you’ll enjoy this as well.