The genetics of inherited vision disorders — notably retinitis pigmentosa (RP) and age-related macular degeneration (AMD) — are an area of intense research. Numerous genes responsible for inherited retinopathies have been identified in the past ten years. At the time of writing, there are at least 242 different genetic disorders, with 202 different genes identified according to the Retinal Information Network (RetNet).
Retinal disorders exhibit an astonishing amount of genetic and phenotypic heterogeneity. Mutations in at least 23 different genes cause autosomal dominant RP, with nearly identical symptoms and disease course. The complexity works both ways: mutations in the ABCA4 gene can cause Stargardt’s disease, cone-rod dystrophy, or RP.
Clinical Diagnostics

Fundus photograph (Pichi et al, J Ophth., 2013)
Because of the growing spectrum of known alleles in disease-causing genes — many of them still of unknown significance — the clinical exam remains important for diagnosis. Classical diagnostic procedures include:
- Fundus photography, which images the morphological appearance of the eyes.
- Electroretinogram (ERG), which measures the electrical responses of various cell types in the retina.
- Electrooculogram (EOG), which measures the resting potential of the retina.
- Pedigree analysis and family history, to determine the probable mode of inheritance.
- Dark adaptation (rod function) and visual field test (central/peripheral vision).
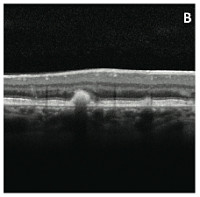
SD-OCT scan of retina (Pichi 2013)
The latest advance in retinal imaging is spectral-domain optical coherence tomography (SD-OCT), which provides a high-resolution cross-section of the retina. With all of these test results in hand, a retinal expert can usually make an accurate diagnosis. Continued visits and monitoring, and possibly examination of other affected family members, provide further confirmation.
Inheritance Patterns
RP and other retinal disorders can be inherited in autosomal dominant, recessive, or X-linked patterns. This Venn diagram, generously provided by Kari Branham of the University of Michigan, shows RP genes associated with different modes of inheritance.

Credit: K. Branham and RetNet
Notably, mutations in at least six genes can cause dominant or recessive disease. There are further complications, such as incomplete penetrance, signs of X-linked disease in carrier females, and even a rare report of digenic inheritance. In other words, even with knowledge of an individual’s genetic variants, it may not be possible to predict whether or not he or she will have the disease.
Incomplete Penetrance
The most striking case is PRPF31, one of the most common dominant RP genes. At least 10% of individuals carrying disease-causing mutations in this gene will not show any symptoms, even late in life. PRPF31, interestingly enough, encodes a component of the spliceosome. The demand for splicing is high in retinal cells, which have lots of tissue-specific isoforms. PRPF31 seems to be the only known gene in which haploinsufficiency causes disease. Interestingly, the carrier-but-unaffected cases exhibit higher innate expression of wildtype PRPF31, likely a compensatory mechanism that prevents disease.
Genetic Testing
Furthermore, genetic testing for predictive purposes isn’t usually necessary, at least for autosomal dominant RP. Because it’s present in every generation, the disorder tends to be a family legacy. It usually manifests by early adulthood, and they know what to look for. By the time that a patient might be consented for genetic testing (i.e. age 18), symptoms of the disease — notably night blindness — are quite evident.
About 40% of patients with a diagnosis of autosomal dominant RP will prove to have mutations in a handful of the most common genes. Perhaps half of the remaining cases harbor occult mutations in a known RP gene. Interestingly, about 8% of the time, a seemingly autosomal dominant family has X-linked disease, most often caused by mutations in RPGR.
That still leaves a fair proportion of individuals with disease caused by an unknown gene. That’s where we, and next-gen sequencing, usually come in.
Future Treatments: Stem Cells and Gene Therapy
Unfortunately, there is no cure for retinitis pigmentosa. The disease usually takes a frustratingly predictable, inevitable course:
- Night blindness and poor dark adaptation, which reflect the rod dysfunction
- Loss of peripheral vision as rods in the peripheral retina die
- Eventually, legal blindness in the late stages of the disease
You can begin to imagine the impact that RP takes on a patient’s quality of life. First there’s the no-seeing-in-the-dark thing, which is problematic throughout life. Then the loss of peripheral vision takes a toll on everyday activities like driving (many patients will lose their license). The loss of central vision often occurs by the age of 40, meaning that a lot of RP patients will spend half of their lives legally blind.
On the bright side, there are some promising therapeutic approaches on the horizon. Gene therapy has shown promise in patients with Leber congenital amaurosis, a form of severe, recessively-inherited retinal degeneration. Mouse models of induced pluripotent stem (IPS) cells have demonstrated the feasibility of regenerating photoreceptor cells and/or retinal tissue. These may be on the horizon, but they’re still a long way off. At best, a distant glimmer of hope for the thousands of patients living with retinal disease.
If you hadn’t guessed already, I have family members affected by RP. Thus, though retinal diseases are interesting and challenging in their own right, there’s another reason I’m so interested. This time, it’s personal.
References
Suma P. Shankar (2013). Hereditary Retinal and Choroidal Dystrophies Elsevier DOI: 10.1016/B978-0-12-383834-6.00147-6