 This week at the American Journal of Human Genetics you’ll find a new method for exome-based mapping and rare variant prioritization in Mendelian disorders. The freely-available software package, MendelScan, is designed to help researchers score and prioritize candidate variants in family exome sequencing studies of Mendelian disease.
This week at the American Journal of Human Genetics you’ll find a new method for exome-based mapping and rare variant prioritization in Mendelian disorders. The freely-available software package, MendelScan, is designed to help researchers score and prioritize candidate variants in family exome sequencing studies of Mendelian disease.
It’s my 50th research publication, and since I’m also celebrating ten years of service at Washington University, it seems like the perfect time for a retrospective.
HapMap Beginnings
I came to work for WashU in 2003, joining the lab of Ray Miller as a bioinformatician. I had a background in computer science and decent Perl programming skills, and enough biology to skimp by. Ray’s lab was in a partnership with Pui Kwok’s group at UCSF to serve as one of the genotyping centers for phase I of the International Haplotype Map (HapMap) project, the ambitious effort to map common genetic variation (SNPs) in human populations. The HapMap is what made all of those SNP chips and genome-wide association studies possible.
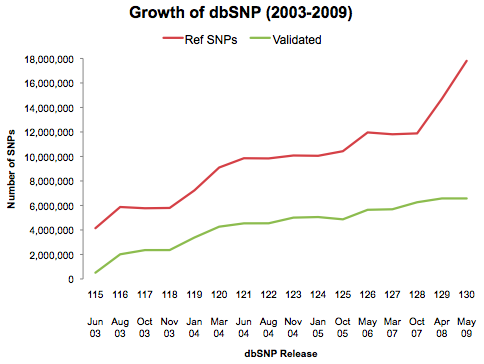
While the HapMap was ramping up, I got my name on a paper for the first time, High-density single-nucleotide polymorphism maps of the human genome (Genomics, 86:2, 2005). SNP discovery was a big deal back then. Simply put, we didn’t have enough SNPs to genotype for the dense map that was planned. Most of dbSNP’s early growth was driven by the needs of the HapMap project.
The HapMap was also my introduction to so-called “big-science”: large-scale, multi-center projects with a data control center and weekly conference calls. They’re a lot of work, but they’ve helped build some of the most important resources for genetic research. I also had my first experiences with Illumina, a company that’s still a big part of my work.
The SNP-in-Primer Effect
Another milestone I can attribute to the HapMap Project was my first first-author paper, Distribution of human SNPs and its effect on high-throughput genotyping, one of the first studies to highlight the connection between nearby SNPs (in primer sites) and genotyping assay failure (including allele dropout).
Mapping C. briggsae
As our role in the HapMap concluded, we took on a new grant: constructing the genetic map of C. briggsae, a small roundworm similar to the well-known model organism, C. elegans. This was a two-part project: first, we used shotgun sequencing data to identify SNPs in a few recombinant inbred lines (RILs). We leveraged the same genotyping platform we’d used for the HapMap (and later, an Illumina GoldenGate array) to genotype the variants in a number of different samples.
It was an interesting project, and I learned far more about worms than I probably ever wanted to. The thing I enjoyed most during this project was the worm research community. They’re a tight-knit and extremely collaborative group. We built some fantastic collaborations, and the three papers out of that one project are a testament to that.
Genetics Hired Guns
We had excess capacity during the C. briggsae project, and that opened the door to some unique collaborations at WashU. Ray knew so many people, both at WashU and in the research community as a whole. Thus, we had a number of smaller projects that helped support the lab and generate publications.
Pharmacogenetics of Warfarin Dose
We worked with Brian Gage and Deepak Voora on the pharmacogenetics of warfarin (coumadin), a widely-used oral anticoagulant. Warfarin is the poster-child for PGx. It has a narrow therapeutic window: too little won’t prevent clotting, and too much causes internal hemorrhaging. The two well-known genes in which genetic variants influence dose are:
- CYP2C9, a cytochrome P450 enzyme that metabolizes warfarin in the liver, and
- VKORC1, a component of the multi-protein VKOR complex involved in the vitamin K cycle that’s targeted by warfarin.
Interesting side note, VKORC1 was first mapped in warfarin-resistant rats, because high-dose warfarin is also used as a rodenticide. We helped with the analysis of targeted 3730 sequencing of some candidate genes in a warfarin patient cohort, and uncovered another gene, (CALU, encoding calumenin) in which variants influenced warfarin dose.
Immunogenetics of Smallpox Vaccination
We also worked with Sam Stanley on a targeted sequencing study to characterize the immunogenetics of smallpox vaccination. Smallpox has been all but eradicated in the West thanks to the development of a vaccine (which most of our parents had). Even so, members of the military were routinely given the vaccine because they might travel to parts of the world where smallpox persisted.
As with many vaccines, the pox vaccine sometimes had side effects, the most common (and measurable) of which was fever. Dr. Stanley’s project sought to identify variants in candidate immune system genes that might contribute to the phenotype.
This was before the completion of the HapMap and the availability of SNP chips, so we had to choose SNPs on a per-gene basis, using dbSNP and preliminary HapMap data. Then we genotyped them in cases and controls, and ran an association study. The study was published in the awesomely titled Journal of Infectious Diseases and highlighted the role of cytokine IL-1A in vaccine response.
The Genome Center
All good things must come to an end, and Ray’s lab eventually closed due to lack of funding. We knew this was coming, so I began looking for a new position at WashU, and I really only had eyes for one place. The Genome Sequencing Center, the place that had helped sequence the human genome while I was still in high school. It was mecca for me. I was fortunate enough to know some people there already, and others at WashU who could put in a good word.
Cancer Genomics
Ultimately, I landed a job in the Medical Genomics Group, charged with the analysis of sequencing data from our high-throughput pipelines. At the time, that was traditional capillary sequencing data. The projects were still ambitious: large-scale targeted sequencing of deadly tumors, like lung cancer and glioblastoma.
AML Cells. Credit: Univ. of Virginia
Things were changing, though. We were using two new sequencing platforms — Solexa and 454 — to unravel the genome and transcriptome of a leukemia patient, one we called AML1. In 2008, it became the first published cancer genome. We went on to publish a second leukemia genome, this one in The New England Journal of Medicine. That was an important milestone for us, because even clinicians and med students read NEJM.
Cancer genomics has been our bread-and-butter for a long time. Collaborations with outstanding oncologists at the Siteman Cancer Center, the Pediatric Cancer Genome Project with St. Jude Children’s Hospital, and our role in The Cancer Genome Atlas, let us work on many of the common cancer types — notably breast cancer and ovarian cancer, as well as brain, colorectal, renal, and others.
The next few years were an incredibly productive period for our group. Next-gen sequencing of tumor genomes was relatively new, and the potential payoff was huge. Admittedly, for many tumor types, the most frequently mutated genes had already been identified by candidate gene sequencing, copy number analysis, and other approaches. NGS nevertheless provided an unbiased assay for genomic changes, and we managed to get some nice publications out of it.
Human Genetics
Our institute took on a number of non-cancer human genetics projects as well. One of the earliest of these was a collaboration with Steve Daiger’s group at the University of Texas, Houston. His group studies inherited retinal diseases, with an emphasis on retinitis pigmentosa (RP), a Mendelian disorder of photoreceptor (rod) degeneration affecting 1 in ~3,500 individuals. I jumped at the chance to work on this project because I have family members with RP. And these were some great collaborators, as evidenced by the publications we managed to put together and our continued interest in discovering new RP genes.
Perhaps because of my background in human genetics and my work on RP, I was pulled into other human genetics projects as they came along too. One of the longest-running of these, a targeted sequencing study of GWAS regions for metabolic syndrome, just came out in PLoS Genetics. Eventually, our workload in human genetics grew enough that we formed a separate analysis group, the group I now manage.
VarSan and NGS Tool Development
Way back during the AML1 project, I offered to try and uncover indels from the 454 data. Given the nature of pyrosequencing, this was not the best idea. But that initial methods development would evolve into VarScan, our tool for SNP and indel calling in individual and pooled samples. It needs to be said that development of analysis tools was not our primary mission. Often there simply wasn’t a good software tool that did what we needed.
Nearly all of our analysis tools were created to meet a specific need in our analyses of NGS data. We developed SomaticSniper to detect somatic mutations in whole-genome sequencing data while allowing for tumor contamination of normal cells that occurs in some types of leukemia. We developed VarScan 2 for somatic mutation and copy number alteration calling in exome data for tumor-normal pairs. The MuSiC package comprised our growing suite of cohort-level mutation analysis tools for cancer. This week’s publication, MendelScan, represents our methodology for linkage-type mapping and scoring candidate variants in Mendelian disorders, most of which was developed during our RP collaboration.
From 50 to 100 Publications
There’s a thrill to getting any paper accepted and published that I enjoy no matter the journal or impact factor. One of the greatest perks of our information age is that biomedical literature goes out to the world, for the entire community to search for, read, and build upon. I also take a certain enjoyment of getting something into a journal that’s never published my work before.
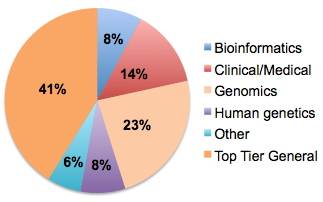
That said, one of the benefits working in big science is taking part of work that can get into top-tier journals. My most-published-in journal by a big margin is Nature (14 papers), followed by the New England Journal of Medicine (4 papers). Those, along with Cell and Science, account for 41% of the publications in my list.
I’ve always liked the notion that 100 publications is the mark of a distinguished research career. Assuming that one works and actively publishes for 20 years, that works out to 5 publications a year. Ten years in, with 50 publications, I’m on track for that. Honestly, I hope it won’t take another decade to reach 100, and I certainly don’t plan on stopping when I do.