
Image credit: CDC Blogs
The rapid adoption of next-gen exome and genome sequencing for clinical use (i.e. with patient DNA) raises some difficult questions about the return of results to patients and their families. In contrast to traditional genetic testing, which usually checks for variants in specific genes, high-throughput sequencing has the potential to reveal a number of secondary findings, i.e., genetic variants with medical relevance but not related to the condition that merited the test.
Two articles in the current issue of AJHG delve into the sticky issue of incidental genetic findings. Holly Tabor et al analyzed de-identified exome data from 6,517 individuals obtained from NHLBI’s Exome Sequencing Project (ESP). They examined the burden of pathogenic variants in three sets of biomedically important genes:
- Genes underlying 31 Mendelian conditions, most of which are inborn errors of metabolism, recommended for newborn screening (NBS, n=39)
- Genes associated with the risk of age-related macular degeneration, a complex disease and the most prevalent form of vision loss (ARMD, n=17)
- Genes known to influence drug response, i.e. replicated pharmacogenetics hits from PharmGKB (PGx, n=14).
Tabor et al, AJHG 2014
Looking only at SNVs called by GATK, the authors identified 10,879 variants affecting the 70 disease genes across the full ESP cohort. Unsurprisingly, filtering this set to include only variants with a high call rate that were listed in OMIM and HGMD for the correct phenotype, reduced the set by over 90%, to around 400 total variants.
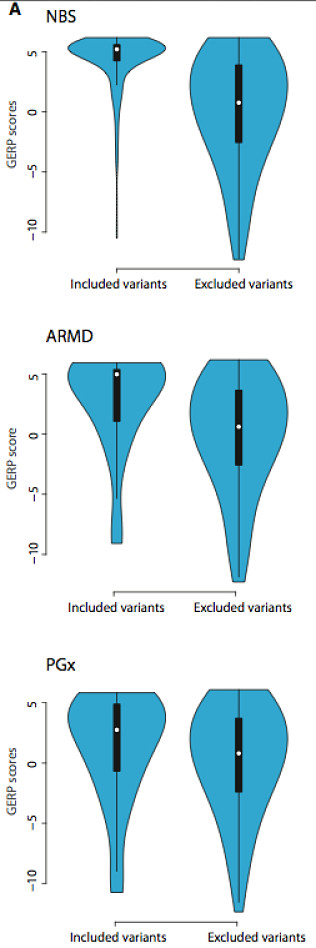
Included versus Excluded Variants
Next, the authors evaluated some of the characteristics of variants that made it through to their final set, versus variants that they’d excluded. Because pathogenic mutations should be under strong purifying selection, one would expect them to be extremely rare and to occur at positions with high conservation across evolution.
The lovely violin plots at right show the mean GERP scores (a measure of conservation) for included versus excluded variants in the newborn screening (NBS), age-related macular degeneration (ARMD), and pharmacogenetics (PGx) genes examined. As the authors hoped, GERP scores were significantly higher for included versus excluded variants, particularly for the severe recessive disease genes screened for in newborns. Included ARMD and PGx variants also had higher GERP scores, but with a wider spread.
A comparison of the Polyphen-2 scores, which offer computational estimates of how damaging amino acid substitutions will be, also showed significant differences, with included variants in the NBS predicted to be far more deleterious than the excluded variants. The effect was again consistent but less striking in the ARMD/PGx sets.
Together, these patterns are consistent with the idea that mutations underlying severe, highly penetrant phenotypes (i.e. the NBS set) are more deleterious — and thus under stronger natural selection — than variants associated with complex phenotypes like ARMD and PGx.
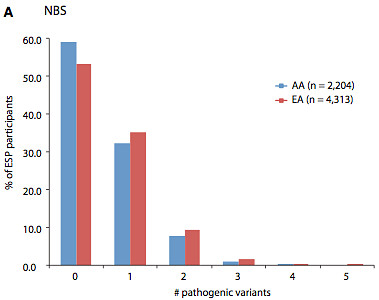
Rate of Incidental Findings
Tabor et al, AJHG 2014
Having established that their final set of ~400 variants was properly vetted, the authors set out to establish the burden of pathogenic mutations that might be found in any individual’s exome. The majority of included variants were rare, with MAF<0.5%.
The carrier burden in the NBS set was surprisingly high (0.57 per exome), with 45% of individuals carrying at least one allele and 11% carrying at least two alleles. If the ARMD and PGx variants were also considered, each individual carried 15.3 risk alleles on average.
These findings challenge the assumption that secondary findings (actionable results) and incidental findings (potential clinical utility) uncovered by exome or genome sequencing are rare. Indeed, a research highlight on the paper from Nature Genetics noted that the study demonstrates the “striking prevalence of actionable incidental or secondary results, including ones of direct clinical usefulness, which might be obtained in patient sequencing.”
In my next post, I’ll tackle the medical community’s opinions about sharing secondary findings, based on a recent survey of 900 genetics professionals.
References
Tabor HK, Auer PL, Jamal SM, Chong JX, Yu JH, Gordon AS, Graubert TA, O’Donnell CJ, Rich SS, Nickerson DA, NHLBI Exome Sequencing Project, & Bamshad MJ (2014). Pathogenic variants for mendelian and complex traits in exomes of 6,517 European and african americans: implications for the return of incidental results. American Journal of Human Genetics, 95 (2), 183-93 PMID: 25087612
[…] RT @laurahercher: A good ‘splainer on new Tabor paper showing a higher than expected rate of sig. incidental findings, from @koboldt http://t.co/hVEIiYWPLA […]