Although I often write about the challenges of next-gen sequencing, it occurred to me that these technologies have reached a certain point of maturity. Thanks to the happy co-evolution of technological and informatics advances, many of the initial problems are essentially solved. Consider the achievements this field has seen over the past several years:
- We developed new algorithms for rapid, accurate mapping of short reads
- Read lengths and base qualities improved dramatically while costs continue to drop
- SAM/BAM formats became community standards for data storage and sharing
- Sequence variants, particularly SNVs, can be detected with high accuracy using NGS data
- Data submission and sharing with appropriate safeguards are now possible in dbGaP.
Perhaps most importantly, sequencing service providers and new benchtop instruments have made NGS available to the wider research community. Nevertheless, some key challenges remain before we can exploit the full potential of high-throughput sequencing.
Finding Samples to Sequence
Once upon a time, the cost and laborious nature of sequencing limited how many samples could be included in a study. Now that throughput is no longer a problem, we are beginning to recognize that samples are the new commodity. Obtaining enough high-quality, properly consented samples remains a significant challenge for our field, particularly when it comes to rare disorders and minority populations.
If every investigator who studied a rare disease provided samples, clinical data, and consent letters to a central collection, we might overcome the sample bottleneck for many diseases. We all know that this will often be unrealistic. Samples have intrinsic value to them. Investigators have careers to build and grants to win. Where we have seen some success is in large-scale, multi-center studies like The Cancer Genome Atlas and the Alzheimer’s Disease Sequencing Project, which bring together investigators and samples and the resources required for some kind of integration.
Compute and Storage for Sequence Data
One of the more obvious challenges of NGS is the sheer volume of data. With sequencing throughputs rapidly outpacing Moore’s Law for compute power (see NHGRI’s timeline of sequencing costs), we find ourselves facing a major CPU and storage problem:
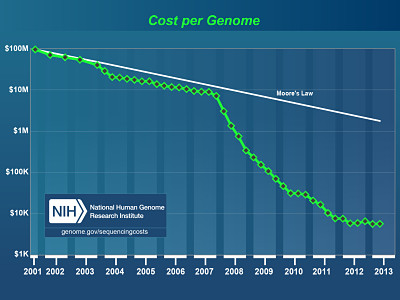
Cost per Genome vs. Moore’s Law (NHGRI)
There have been many innovative algorithmic developments that help address the growing data-to-CPU ratio. Storage, however, is another matter. Without losing information, storing aligned next-gen sequencing data takes disk space. The more data you have, the more space required.
How long should we store NGS data? Storage isn’t free — it requires hardware and maintenance and physical space — so at some point (especially given shrinking research budgets), we will need to address this issue.
Indels, SVs, and Other Difficult Variants
Detection of SNVs in NGS data for individuals or tumor-normal pairs is largely a solved problem, but that can’t be said for other types of variation. You know the kinds of variants I’m talking about: insertions, deletions, duplications, inversions, and more complex rearrangements are tough to characterize using short-read sequencing data. Many impressive algorithms have come out to detect and characterize these, but they all suffer much higher false-positive and false-negative rates than we see for SNVs.
It is possible that the current paradigm for high-throughput sequencing data — paired-end reads in the 100-250 bp range — may never yield an optimal solution for detecting these kinds of variants. At least, I think it’s more likely that some of the long-read technologies racing to market stand a better chance at letting us characterize these variants with >90% accuracy.
Discovery and Interpretation
Next-gen sequencing technologies have opened the floodgates for genomic data. Thousands of whole genomes and hundreds of thousands of exomes have been sequenced already. You will notice, however, that the rate at which new genomic discoveries are made (published), while impressive, does not match the rate at which new samples are sequenced. In other words, we have far more data than findings.
Previously I wrote about some of the reasons exome sequencing studies can fail even for well-characterized, monogenic Mendelian disorders, and the need for functional validation of genomic findings before we accept them as fact. Our ability to find, associate, and implicate genetic variants and candidate disease genes far outstrips our ability to understand them.
There is a pressing need for better downstream analysis tools to help interpret genomic data, especially in the ~97% of the genome that lies outside of the exons of known protein-coding genes.
Clinical Translation of Genomic Discoveries
Perhaps more importantly, the findings enabled by next-gen sequencing must eventually translate into improvements to human health. That, essentially, is our funded mandate.
There are many ways to make this happen. Identification of new disease genes may provide new therapeutic targets, and improve the predictive abilities of genetic testing. Clinical sequencing of patients suffering from disease may eventually guide diagnosis and treatment decisions.
Importantly, these pathways to clinical translation will require the expertise in many disciplines: molecular biology, pharmacology, genetic counseling, clinical care, and many others.
We “genomics” people can’t do it alone. Reaching our ultimate goal will probably require ambitious multi-disciplinary collaborations focused on specific health problems. Genomics and NGS will certainly be a part of that, but not the biggest part. Not even half.
[…] genetically modified food. How Many Ants Live in New York City? Are your data normal? Hint: no. Remaining Challenges of Next-Gen Sequencing Whooping Cough Linked to Shorter Life Expectancy Bioinformatics is not something you are taught, […]