Exome sequencing has undeniably transformed the study of rare inherited disorders, enabling the rapid identification of hundreds of new diseases genes in the past few years and spurring the adoption of clinical exome sequencing as a frontline diagnostic tool. That’s great news. Hooray for the exome!
Is it a fantastic discovery tool? Absolutely. But it’s not a magic bullet.
The less-publicized outcome of widespread exome sequencing is that “hit rate” — the proportion of sequenced cases for which a likely genetic cause is found — has largely remained the same. For most studies, it’s in the neighborhood of 40-60%. Higher success rates have been reported, but these usually involve cherry-picking cases or the inclusion of patients who who’d not undergone any a priori molecular testing.
The bottom line is that a significant fraction of rare disease cases fail to achieve a genetic diagnosis by exome sequencing. When this happens, it’s tempting to consider whole-genome sequencing as the logical next step. Yet it’s hard to know how often that will help. A new study in the American Journal of Genetics has begun to answer that question.
Keren J. Carss et al performed exome sequencing, genome sequencing, or both on 722 patients with inherited retinal disease. IRDs offer a number of advantages for studies like this due to exceptional phenotypic, genetic, and allelic heterogeneity. There are more than 250 known genes associated with IRDs, and they can be inherited in every possible mode. Dominant, recessive, X-linked, and even mitochondrial inheritance have been documented.
Cohort Phenotype Composition
The 722 cases described here were recruited under the NIHR BioResource Rare Diseases research study. That’s in the United Kingdom, by the way. So are most of the study’s authors, a fact made plain by the UK epidemiology figures and the reference to Genome England as an example of a large-scale genome sequencing initiative in the Introduction. The phenotype composition generally reflects the prevalence of inherited retinal diseases:
- 311 had retinal pigmentosa (RP), characterized by night blindness and progressive rod photoreceptor loss.
- 101 had “retinal dystrophy”, a broader term that could mean RP or other retinal degenerations
- 53 had cone-rod dystrophy, which affects cone photoreceptors, causing loss of color and perceptive vision.
- 45 had Stargardt disease, the most common form of inherited juvenile macular degeneration.
- 37 had macular dystrophy, a broader term for diseases affecting the macula, the central portion of the retina.
- 37 had Usher syndrome, a condition characterized by vision loss (RP) and hearing loss.
Genome and Exome Sequencing
Too often, I see a paper with “Whole-genome sequencing” in the title in which a handful of samples actually obtained WGS, and the rest got targeted sequencing. Although I understand the economics of such a design, it feels like a bait-and-switch. This study did not disappoint: 650 of 722 cases underwent WGS, with the remaining 72 getting exome only. The average depth for WGS was 37x, which is standard, but the average exome depth (43x) is a little low. That’ll be relevant in a minute.
The bioinformatic analysis and interpretation strategies look solid. The authors searched for high-quality rare coding variants in a curated set of 224 retinal disease genes. Candidate causal variants were reviewed in IGV, and assessed in the context of databases (like HGMD), segregation, and how well the clinical phenotype matched the phenotype associated with the gene.
Pathogenic Variants Detected
They identified a likely causal variant in 404 individuals (56%). That’s slightly on the high end of realistic success rates, but as the authors admit, 152 individuals in the cohort had had no prior genetic testing (and 63% of them were solved). The hit rate also varied by phenotype. RP, the largest phenotype group, saw a hit rate of 54% which is right where we expect it to be.
The success rate varied widely for other phenotypes, ranging from 29% in cone-rod dystrophy to 84% of Usher syndrome (the latter is not terribly surprising, since variant interpretation is arguably the easiest for rare, recessive conditions).
Solve Rates Varied by Ancestry
A particularly intriguing observation was that diagnostic success varied by individual ancestry. The success rate was considerably lower for individuals of African ancestry (30%) compared to individuals of European (57%) or South Asian ancestry (53%). I admired how the authors remarked:
Higher genetic diversity in African populations, combined with underrepresentation of non-European populations in control datasets, result in an excess of rare and apparently rare variation in these individuals, rendering variant interpretation more challenging.
Another intriguing ancestry tidbit was that 66% of pathogenic variants in South Asian cases were homozygous, compared to 18% of pathogenic variants in European Americans. The authors argue that this is likely due to greater consanguinity in South Asian populations, which may also explain why their hit rate was comparable to that of European-ancestry individuals despite underrepresentation in control databases.
Exome and Genome Performance
Some 117 individuals underwent exome sequencing first, and in 59 of those (50%), a likely causal variant was uncovered in this first pass. Next, the authors selected 45 of the 58 exome-negative individuals for whole genome sequencing.
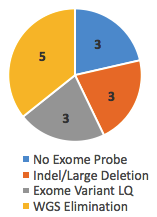
Reason Variant Was Missed
Of these, 14 cases, or 31%, achieved a genetic diagnosis after WGS. But take note of the reasons those variants were missed. Three of them had no probe (and thus no coverage) in the Nimblegen v3 exome kit and 3 were large indel/deletions missed by exome sequencing. Another 3 were called in the exome but flagged as LQ (low quality), likely due to poor coverage or representation of both alleles. In these 9/45 cases (20%), WGS did succeed where exome failed.
Yet the remaining 5 variants were called in the exome, but not considered causal until WGS eliminated all other possibilities. Should these go in the win column for WGS? I’m not sure. If they aren’t, then the true discovery rate for WGS in exome-negative cases in this study is 20%.
Whole-genome Advantages
Although the numbers are modest, whole-genome sequencing undoubtedly enabled the researchers to uncover more pathogenic variants in these cases. A wonderful example is offered in Figure 1:
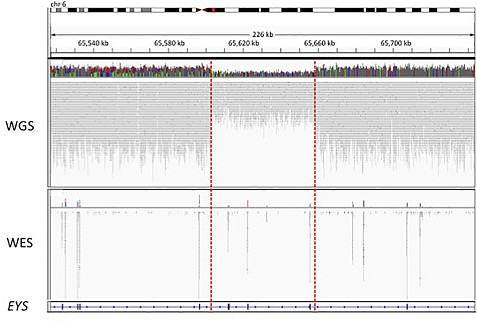
Carss et al, AJHG 2017
In this case, a patient with recessive RP had one pathogenic variant (a missense change in EYS) detected by exome sequencing. However, it took whole-genome sequencing to identify the second disease allele, a heterozygous ~55 kb deletion spanning at least three other exons in the gene.
Pathogenic Noncoding Variants
The authors also leveraged our current knowledge of gene-phenotype relationships, and the comprehensive nature of WGS data, to identify three pathogenic noncoding variants. All of these where deep intronic variants that likely affect splicing, and were found in patients whose phenotypes corresponded to defects of a specific gene:
- In 16 individuals with Stargardt disease, caused by recessive-acting variants in the ABCA4 gene, the authors identified a rare intronic variant. It was homozygous in two cases, compound-heterozygous with a coding variant in nine, and the only ABCA4 variant in the remaining 5 (which are classified as “partially solved” cases).
- In a patient with Usher syndrome, the authors identified a known pathogenic noncoding variant (intronic) that causes the retention of a pseudo-exon.
- In two unrelated males with choroideremia, an X-linked disorder caused by mutations in the CHM gene, the authors uncovered a novel deep intronic variant creates a cryptic splice site that causes retention of a 224-bp cryptic exon.
For all three regulatory variants, the investigators knew where to look because the patient’s phenotype strongly pointed to a known gene. This is a clever strategy for beginning to tease out regulatory variation in Mendelian disorders, and may help open the door for even more discoveries.
In Summary
This was a well-written paper that showcased some of the advantages to whole-genome sequencing over exome sequencing for uncovering the genetic basis of rare diseases. I hope (and expect) we’ll see more studies like it as WGS becomes ever more practical to apply as a frontline diagnostic tool.
[…] date, the use of this technology has resulted in about 40% of the previously unexplained cases being […]