Understanding the genetic basis of common disease is an important goal for human genetics research. Nothing that we do is easy — the ~25% success rate of exome sequencing in monogenic (Mendelian) disorders is proof enough of that — but the challenges of complex disease genetics are considerable.
Cardiovascular and metabolic diseases in particular arise from a complex array of factors beyond genetics, such as age, diet, and lifestyle. We also expect that most of the genetic variants conferring risk will have small effect sizes, which makes their identification all the more difficult.
Common Variation: the GWAS
We do have some powerful tools. Over the last decade, researchers have leveraged high-density SNP array genotyping — which is relatively cheap, high-throughput, and captures the majority of common genetic variation in human populations — to conduct massive genome-wide association studies (GWAS) of common disease.
This approach has yielded thousands of genetic associations, implicating certain loci in the risk for certain diseases.
Rare Variation: Sequencing Required
Yet the variants identified (and genes implicated) explain only a fraction of the genetic component of these diseases, and they generally don’t interrogate rare variation, i.e. variants with a frequency of <1% in the population. The only way to get at these is by sequencing, and the rapid evolution of next-generation sequencing technologies has begun to make that feasible.
A new study in Nature describes such an effort: a search for rare variants associated with risk for myocardial infarction (MI), or in layman’s terms, heart attack. It not only yielded some key discoveries, but showcased some of the challenges and expectations we should have in mind when undertaking large-scale sequencing studies of common disease.
NHLBI’s Exome Sequencing Project
A few years ago, the National Heart, Lung, and Blood Institute of the NIH did something very wise: they funded a large-scale exome sequencing project (referred to by many as “the ESP”) comprising several thousand samples from a number of cohorts. As one of the earliest widely-available exome sequencing datasets at this scale, the NHLBI-ESP quickly became an important resource for the human genetics community.
At the most basic level, it tells us the approximate frequencies of hundreds of thousands of coding variants in European and African populations. Unlike the 1,000 Genomes Project, however, the ESP collected deep phenotyping data, enabling genetic studies of many complex phenotypes.
First Pass: Association and Burden of Rare Variants
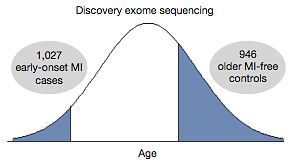
Discovery phase: case/control selection (R. Do et al, Nature 2015)
Ron Do and his 90+ co-authors designed a discovery study for the extreme phenotype of early-onset MI. Across 11 studies in the ESP, they identified 1088 individuals who’d had a heart attack at an early age (<50 for men, <60 for women). As a control group, they selected 978 individuals who were at least a decade older than that but had had no heart attack. And with the exome data already in hand, they could search for rare variation associated with the phenotype (early-onset MI) in different ways:
- Individual variant associations. Among low-frequency (MAF 1-5%) coding variants, no single variant was significantly associated with the phenotype.
- Gene-based associations. Rather than considering individual variants, the authors looked at the “burden” of rare variants at the gene level. For each gene, the authors compared the fraction of samples with at least one rare (MAF<1%) coding variant between cases and controls. No genes had significant associations.
Importantly, gene-based association tests (also called “burden tests”) can be performed in a variety of ways. What frequency threshold should be used? What distinguishes a benign variant from a damaging one? The authors set a MAF ceiling of 1% and considered three sets of variants:
- Nonsynonymous. All missense, splice site, nonsense, and frameshift variants.
- Deleterious. The nonsynonymous set above, minus missense variants predicted to be benign by Polyphen2.
- Disruptive. Nonsense, splice site, and frameshift variants only.
These were reasonable choices, comparable to what we or other groups do in this kind of study. Still, there were no significant results so it was on to phase 2.
Genotype Imputation and Exome Chip
PCA analysis (R. Do et al, Nature 2015)
It’s very possible that there are individual variants and genes associated with the phenotype, but the authors didn’t examine enough samples to find them (by their own calculations, in a best-case scenario the power for a study of this size was about 0.2).
So they pursued a few strategies to increase the sample numbers substantially. Across the 11 cohorts there were over 25,000 early-onset MI cases (and an even larger number of suitable controls) but these samples only had SNP array data, and the vast majority of markers on SNP arrays are non-coding.

Low freq. variant follow-up (R. Do et al, Nature 2015)
So the authors undertook a major effort to impute (statistically predict) the genotypes of 400,000 coding variants based on the SNP array data and a reference panel of samples that had both SNP array and exome data. This was a herculean effort that only merited two sentences in the main text (there are severe restrictions on a “letter” to Nature) because it yielded no finding: no significant association, even with imputed genotypes for 28,068 cases and 36,064 controls.
The authors also performed high-throughput genotyping with the so-called “exome chip,” which looks at ~250,000 known coding variants, in about 15,000 samples. At the time, the cost of running that many exome chips likely exceeded $1 million. Yet there were no significant associations, so that, too, got 2 sentences in the main text.
Targeted Resequencing Follow-up

Sequencing follow up (R. Do et al, Nature 2015)
The authors needed sequencing data, but they also needed more samples. These things not being free, they decided to choose six of the most promising genes (based on not-entirely-disclosed biologic / statistical evidence) for targeted resequencing in about 1,000 more samples. Once that was done, and the analysis performed yet again, of those (APOA5) looked promising. So the authors sequenced just that gene in three additional studies. This was a mix of PCR-based 3730 sequencing and multiplexed long-range PCR libraries on a MiSeq instrument.
Finally, after sequencing the exons of APOA5 in 6,721 cases and 6,711 controls, the authors had an association that reached genome-wide significance: 5 x 10-7 when a burden test with all nonsynonymous variants was used (the threshold is 8 x 10-7).
More Exome Sequencing Yields Most Obvious Gene Ever
The fourth and final follow-up strategy was simply to do more exome sequencing of ~7,700 individuals, bringing the total to 9,793 samples (4,703 cases and 5,090 controls). After applying a variety of burden test strategies, the authors found exactly one gene with significant evidence of association: LDLR, which encodes the low-density lipoprotein receptor. It’s been known for many years that mutations in LDLR cause autosomal-dominant familial hypercholesteremia, and high LDL cholesterol is one of the top risk factors for MI, so this is both a biologically plausible and completely unsurprising hit.
About 6% of cases carry a nonsynonymous variant in LDLR, compared to 4% of controls, so the odds ratio is about 1.5. This is a classic GWAS result, isn’t it? Very obvious candidate gene achieves statistical significance and the odds ratio is very low.
Interestingly, however, if the authors apply more stringent criteria for variants, the effect becomes more dramatic:
- Deleterious variants (i.e removing Polyphen’s benign missense) were in 3.1% of cases, 1.3% of controls, yielding the best p-value 1 x 10-11 and an odds ratio of 2.4.
- Strictly-deleterious missense (requiring 5/5 programs to call a missense variant deleterious) were in 1.9% of cases and 0.45% of controls, yielding a slightly higher p-value of 3 x 10-11 but an odds ratio of 4.2.
- Disruptive variants had the highest odds ratio (13.0), but with a much higher p-value (9 x 10-5) and affecting just 0.5% of cases. These are basically familial hypercholesteremia carriers.
Conclusions
At first glance, one might wonder how this came to be a Nature paper because there were no truly novel findings. LDLR was already well known, and APOA5, which encodes an apolipoprotein that regulates plasma triglyceride levels, was already a strong candidate gene for MI. In fact, two other genes related to APOA5 function had already been reported for association with plasma TG levels and early-onset MI, and the gene resides in a known locus for plasma TG levels identified by classic GWAS.
True, that region had extensive LD and it wasn’t clear which of the few genes there were involved in the phenotype. And technically, APOA5 had not yet been established as a bona-fide gene for early-onset MI. This is the final nail in the coffin, but look what it took to get here: exome sequencing, followed by targeted sequencing, and then even more targeted sequencing. It’s glossed over in the paper, but every step in the authors’ pursuit of APOA5 required timely, careful analysis of the genetic evidence.
In the last part of their letter, the authors discuss some of their power calculations for large-scale genetic studies of this nature. They sought to answer that pivotal question, “How many samples do we have to sequence to find something?”
Because of the challenge of distinguishing benign from deleterious alleles, and the extreme rarity of the latter, well-powered studies of complex disease will require sequencing thousands of cases. Here’s the authors’ power calculations for a gene harboring a median number of nonsynonymous variants:
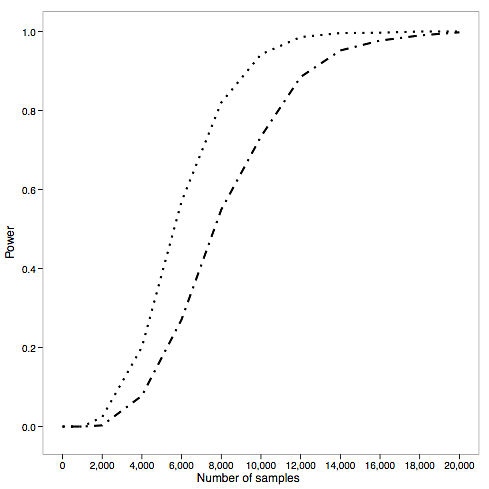
Power to detect gene with median # of variants (R. Do et al, Nature 2015)
- In a best-case scenario — a gene harboring large numbers of nonsynonymous variants, each conferring the same direction of effect — we’re talking 7,500 samples to achieve >90% power.
- In a more likely scenario (i.e. the power calculations above) — a gene harboring median numbers of nonsynonymous variants — it’s 10,000 or more samples.
Generating, managing, and analyzing exome or genome sequencing data for these sample numbers is a massive undertaking. Undoubtedly, this will be the mission for us and other large-scale sequencing centers for years to come.
References
Do R, Stitziel NO, Won H, Jørgensen AB, Duga S, et al (2014). Exome sequencing identifies rare LDLR and APOA5 alleles conferring risk for myocardial infarction. Nature PMID: 25487149
My fear is that, unless conducted in an unbiased way and with 10K+ samples as you pointed out, sequencing studies like targeted resequencing might just be a fancy way of wrapping candidate gene studies that lead to little insight at the cost of a lot of confusion.
Great write up.
I don’t really care if they found rare variants or not – that’s science. What I do care about is that they make their analyses reproducible, which they didn’t.