 If you compare any individual’s genome to the human reference sequence, you’ll find around 3 million differences. Most of these (95%) area already known, and have been catalogued in databases like dbSNP. Many are common, and shared by 5% or more of human populations. They may still have biomedical relevance, of course; genome-wide studies of common genetic variation (GWAS studies) have found thousands of genetic loci associated with disease susceptibility and other complex traits.
If you compare any individual’s genome to the human reference sequence, you’ll find around 3 million differences. Most of these (95%) area already known, and have been catalogued in databases like dbSNP. Many are common, and shared by 5% or more of human populations. They may still have biomedical relevance, of course; genome-wide studies of common genetic variation (GWAS studies) have found thousands of genetic loci associated with disease susceptibility and other complex traits.
But there are still huge numbers of rare (MAF<0.5%) and low-frequency (MAF<5%) genetic variants. Their contribution to human health is harder to understand, particularly because such variants:
- Are usually not included on high-density SNP arrays
- Occur in few individuals, and thus require large cohorts
- Have low individual power for genetic association
One way to address the challenges of rare variants is to study them in founder populations in which such variants are more common. Ashkenazi Jews and Amish families, for example, have undergone population bottlenecks effects: a limited number of founders gave rise to the current populations.
This breeding isolation, whether cultural or geographic in nature, increases the frequency of some variants that are otherwise quite rare in broad populations. And if those variants underlie a genetic disorder, the risk of the disease is increased. Ashkenazi Jews, for example, have increased risk of many uncommon genetic disorders.
Finland has a unique population history — a bottleneck followed by geographic isolation — the result of which is a Finnish “disease heritage”: a high incidence of 40+ Mendelian disorders. Dozens of rare Mendelian disease genes were mapped in Finns, and that knowledge is valuable for understanding disease biology. What about rare variants underlying common, complex disease? Here the Finns have an important resource: nationalized health records with decades of follow-up data.
Sequencing Initiative Suomi (SISu)
The Sequencing Initiative Suomi (SISu) aims to combine the unique population structure, the health records, and the substantial Finnish interest in genetics. The first study from SISu, just out in PLoS Genetics, compares the exomes of 3,000 Finns to an equal number of non-Finnish Europeans (NFEs). They found:
- A depletion of “singletons” (variants only seen in one individual) in Finns: 3.7 times fewer singletons than NFEs
- An excess of low-frequency variants (MAF 0.5-5%) in Finns relative to NFEs
- Similar patterns of common variants between Finns and NFEs
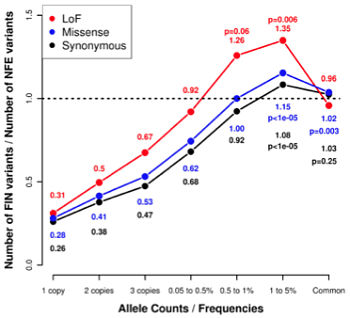
Lim et al, PLoS Genetics 2014
All of these are consistent with the expected bottleneck effect on Finnish populations. When variants were stratified by annotation (i.e. their predicted effect on genes), Finns had a higher proportion of likely-deleterious missense variants and more severe loss-of-function (LoF, or protein-truncating) variants. The average Finn had 0.160 homozygous LoF variants, whereas the average NFE had 0.095.
To determine if some of these enriched LoF variants have phenotypic effects, the authors genotyped 83 of them in 36,262 individuals from three large Finnish cohorts. Using the deep phenotype data — quantitative traits like blood pressure, lipids, etc. — they found 5 significant associations.
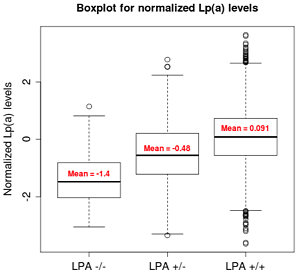
LPA level (Lim et al, PLoS Genet 2014)
One of these was an association between splice site variants in the gene encoding lipoprotein A (LPA) and decreased levels of circulating lipoprotein A. As it happens, circulating LPA is a risk for coronary heart disease. Looking at the medical records showed that LPA splice variants are protective for cardiovascular disease.
This is only a proof-of-principle study, the tip of the SISu iceberg. Yet it shows the value of sequencing Finnish populations to identify rare variants contributing to complex diseases. Undoubtedly, as large-scale sequencing of Finnish cohorts continues at places like WashU and the Broad Institute, we’ll have even more power to identify genes relevant for common diseases.
Very interesting!
If I understand the methods correctly, it seems that these are different individuals than those included in 1,000 Genomes. Am I right?