GoNL Consortium, Nat. Gen. 2014
The last time I checked, the database of human genetic variation (dbSNP) contained over 50 million unique sequence variants. And yet, as anyone who analyzes exome or whole-genome sequencing data can tell you, every individual harbors a significant number of variants (usually around 5% of single nucleotide variants, or SNVs) that dbSNP has never seen.
These “private” or rare variants undoubtedly contribute to important phenotypes, such as disease susceptibility. Non-SNV variants, like indels and structural variants, are also under-represented in public databases. The only way to fully elucidate the genetic basis of a trait is to consider all of these types of variants, and the only way to find them is by large-scale sequencing.
In this month’s issue of Nature Genetics, the Genome of the Netherlands (GoNL) Consortium reports the whole-genome sequencing of 250 Dutch families from 5 biobanks across the Netherlands. The families comprised mostly parent-child trios (n=231), along with some family quartets with monozygotic (n=11) or dizygotic (n=8) twins. All told, it was 769 individuals whose genomes were sequenced ~13x depth.
Variant Calling
Granted, this is a modest coverage depth, considering that sequencing to 30x or 40x might be considered standard. To help address this, the authors performed joint sample calling with GATK. That, along with a combination of 10 indel/SV calling tools, yielded the following:
- 20.4 million biallelic SNVs
- 1.2 million biallelic indels of 1-20 bp
- 27,500 larger deletions (>20 bp)
Here’s a quick tour of the highlights in each variant class
Single Nucleotide Variants (SNVs)
GoNL Consortium, Nat. Gen. 2014
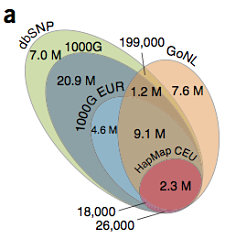
Half of the 20.4 million SNVs discovered in this study were rare, with MAF < 0.5%. The others were not quite evenly divided between low-frequency SNVs (4.0 million with MAF 0.5-5%) and common SNVs (6.2 million with MAF >5%). Altogether, there were around 7.6 million SNVs that were novel to dbSNP 137. Most of those 75%) were singletons, meaning that they were observed in just one individual. If we consider only the 500 unrelated individuals sequenced (the parents), that’s about 15,200 novel contributed variants per sequenced genome.
Among the ~2 million singletons uncovered in the European panel of a different project (1,000 Genomes), 16.5% were observed in GoNL. The authors therefore expect that a “substantial number” of singleton variants reported by these projects will be seen again as larger European cohorts are sequenced. Even so, that’s a lot of “private” variation. Remember, too, that these cohorts are from northwest Europe, arguably one of the best-characterized ancestry groups thus far.
Indels and Structural Variants
Compared to SNV calling, the detection of indels and larger SVs remains a considerable challenge. Anyone working in NGS informatics can tell you that. The authors have put forth a good effort in this arena by combining the results of 10 different variant callers: GATK UnifiedGenotyper, Pindel, 1-2-3SV, Breakdancer, DWAC, CNVnator, FACADE, MATE-CLEVER, GenomeSTRiP and SOAPdenovo. These are all different algorithms, but they boil down to five approaches for uncovering indels and SVs:
- Gapped reads alignments to the reference (e.g. GATK)
- Split reads, an approach pioneered by Pindel
- Paired-end read distance/orientation (e.g. BreakDancer)
- Overall read depth changes (e.g. CNVnator)
- De novo assembly of SV breakpoints.
GoNL Consortium, Nat. Gen 2014
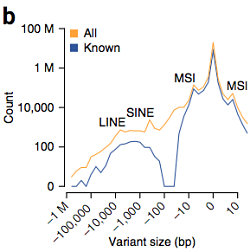
Some of the tools use one approach, while others employ multiple approaches. No single indel/SV caller has emerged as vastly superior to all others, so combining the results from a suite of different tools seems like a good strategy. The authors divided variants into three size categories (1-20 bp, 20-100 bp, and >100 bp) and kept any SV detected by at least two orthogonal tools. Their validation rate (138/144, or 96.5%) for randomly-chosen SVs of at least 20 bp is impressive.
The size distribution of consensus calls showed peaks at +/- 4 bp (microsatellite instability), ~300 bp (SINEs), and ~6 kbp (LINEs). Not remarked upon in the manuscript is the largest peak right near zero, since 1-2 bp indels are by far the most common. While 54.4% of short indels (<20 bp) were already in dbSNP, virtually all of the mid-size deletions (30-500 bp) were not (98.4%). Thus, this study helps fill an important gap in the catalogue of human sequence variation.
Functional Variation
Rare, low freq, and common variant distributions (GoNL, Nat. Gen 2014)
Because these families were not obtained “on the basis of phenotype or disease,” their patterns of genetic variation provide a useful model for apparently healthy individuals.
Rare Loss-of-Function Variants
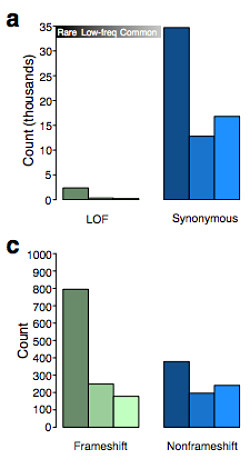
Among rare variants identified in this study, the authors observed an excess of nonsense SNVs and frameshift indels, consistent with the expectation that damaging variants would be under strong purifying selection.
A similar excess-of-rare-events was evident for larger deletions that removed the first exon or >50% of the coding sequence of a gene. The effect was even stronger when considering only genes in the OMIM database, reflecting strong purifying selection against structural changes in key genes.
On average, each individual in GoNL had about 60 nonsense or splice-site SNVs. Most of these, however, were common in the cohort (MAF>5%, and thus unlikely to be deleterious), which illustrates the need for cautious interpretation of apparent loss-of-function (LOF) variants. Looking at rare variants, and using synonymous SNVs as a baseline, the authors estimate that each individual might have 4-5 rare loss-of-function SNVs.
Compound Heterozygous Events
Individuals that were compound-heterozygous (i.e. one variant on each parental haplotype) for rare loss-of-function SNVs/indels/SVs were extremely rare. The authors found just 3 such instances across the cohort (an average of 0.01 events per individual). Such events are thus of considerable interest for disease studies.
Compound heterozygosity for common LOF variants should have been far more prevalent, because these are less likely to be truly deleterious. Indeed it was; there were about 3 compound heterozygous events of common LOF variants per individual. Interestingly, the 1,917 such events observed across the entire cohort were confined to 11 genes (C11orf40, DEFB126, GSTT2, HTR3D, KRTAP4-8, MS4A14, OR13C2, SIGLEC12, TRY6, VWDE, and WNK1) which all seem to have high mutation tolerance.
HGMD False Positives
The human gene mutation database (HGMD) is a commercial repository of “disease causing” variation in humans. When the authors of this study annotated variants with HGMD information, each individual harbored about 20 such variants. In other words, HGMD annotation would suggest that a large number of GoNL individuals have diseases with profound physical (or even lethal) consequences. Whoops.
It’s possible that the HGMD variants simply cause disease in non-Dutch populations, or have low penetrance. An alternative possibility is that HGMD has a lot of false positives. Among the 1,093 HGMD variants in GoNL, almost a third had MAF>1%, which is much higher than the frequency of the diseases they’re reported to cause.
De Novo Mutations
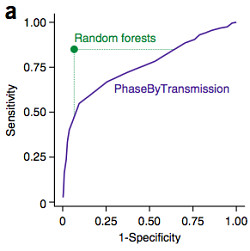
De novo mutation calling performance, GoNL, Nat. Gen 2014
One of the most fascinating aspects of this study was the exploration of de novo mutations (variants present in a child but absent from both parents). These events are extremely rare (occurring at a rate of around 1 in 100 million bases), and identifying them absolutely requires sequencing at least three genomes: an individual and both biological parents.
Even then, they’re very difficult to find: Across the 258 independent offspring in GoNL there were 4.5 million apparent Mendelian violations. The authors applied a method (PhaseByTransmission) to refine this to around 29,162 candidate autosomal de novo mutations. That’s about 63 per offspring, far too many.
So the authors attempted to independently validate over 1,000 candidate de novo mutations by orthogonal sequencing, and found that around 50% were false positives. Some independent Complete Genomics data for 19 parents and 1 child revealed another 1,137 events that were false positives. From these 2,270 observations, the authors developed a random forest classifier to predict whether a predicted mutation would be truly de novo or not based on a number of different properties. This is something that many other groups (including ours) have done for somatic mutation calling in cancer genomes.
The classifier in this study, which had an estimated accuracy of 92%, relied primarily on factors related to the sequencing depth and read counts, which happens to be the basis for mutation detection with VarScan 2. When applied to the GoNL dataset, the classifier nominated 11,020 high confidence de novo mutations — roughly 42.7 per offspring — with a range of 18 to 74 per offspring. That’s still a bit higher than it should be, but still reasonable for downstream analysis.
Paternal Age and de novo Mutation Rate
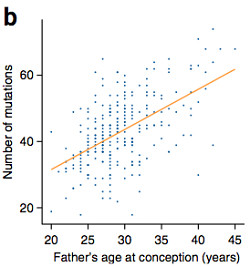
GoNL Consortium, Nat. Gen 2014
The authors observed a significant correlation between the father’s age at conception and the number of de novo mutations in the child. This is the third study to report such a trend, and the largest sample size yet. Although the ages of mother and father are highly correlated, its effect on de novo mutation rate was primarily due to paternal influence.
The authors estimate that each additional year of paternal age caused a 2.5% increase in the number of de novo mutations in the child. Under their model, about 75% of de novo mutations come from the father, and 25% from the mother. Phase analysis using read pairs (a complex process I won’t go into) revealed that 76% of de novo mutations were indeed on the paternal haplotype. So you can thank your dad for 3/4 of your de novo mutations.
De Novo Indels and SVs
The authors attempted to find de novo indels and structural variants. It didn’t go well.
In Summary
The authors have employed moderate-coverage whole genome sequencing to build a resource of 1,000 haplotypes for a small, densely-populated country in northwestern Europe. They added 7.6 million SNVs to dbSNP, and also characterized a large number of new indels and SVs. Many more studies which apply genome sequencing to large population cohorts will be necessary to fill out the catalogue of human genetic variation.
[…] One example is the Dutch GoNL project, which was recently published and discussed by Dan Koboldt on massgenomics.org. In summary, the mutation spectrum of recessive disorders is unknown and databases are of limited […]