 This week at the Advances in Genome Biology & Technology meeting in Florida, we’ll undoubtedly hear more about Illumina’s new NGS platforms, particularly the HiSeqX Ten. Illumina claims that this $10 million factory installation will permit the sequencing of human whole genomes per year at under $1,000 each (equipment and reagents cost).
This week at the Advances in Genome Biology & Technology meeting in Florida, we’ll undoubtedly hear more about Illumina’s new NGS platforms, particularly the HiSeqX Ten. Illumina claims that this $10 million factory installation will permit the sequencing of human whole genomes per year at under $1,000 each (equipment and reagents cost).
To achieve this economy, however, every HiSeqX Ten installation must do 15,000 genomes per year. For four years, which means 60,000 samples. Remember, the licensing terms for the technology specify that it can only be used for whole-genome sequencing, no exomes, no targeted panels.
If we ignore the elephant in the room — the logistics of finding, consenting, and funding that many samples — we might expect that tens of thousands of human genomes will be sequenced in the coming years.
As a bioinformatician, I have the unfortunate duty of informing you that we haven’t yet reached the point you saw in GATTACA: no machine yet exists to predict someone’s entire medical future from the readout of their genetic code. There are, however, some low-hanging fruit: promising applications of large-scale human genome sequencing which are likely to yield results.
1. Unsolved Mendelian Disorders
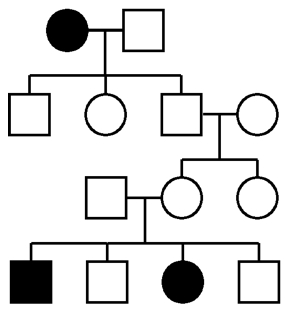 When exome sequencing became widely available, Mendelian disorders were among the first things tackled. Rare, highly penetrant disorders with multiple affected samples that have already been screened by exome sequencing might be the best place to start. Remember, the success rate for exome sequencing of rare genetic disorders hovers at around 30% overall.
When exome sequencing became widely available, Mendelian disorders were among the first things tackled. Rare, highly penetrant disorders with multiple affected samples that have already been screened by exome sequencing might be the best place to start. Remember, the success rate for exome sequencing of rare genetic disorders hovers at around 30% overall.
There are several reasons such studies can fail, but one is simply that it’s not whole-exome sequencing. At least 5-10% of coding bases don’t achieve reliable coverage by that approach. Whole-genome sequencing is the obvious next step.
2. Inherited Pediatric Disease
The birth of a child with a congenital disorder often marks the start of a years-long diagnostic odyssey. Newborn genetic testing varies widely from state to state in the U.S. and captures only a small fraction of the well-established inherited disorders. Laboratory tests, karyotyping, CGH arrays, and candidate-driven genetic testing are often frontline approaches.
This is an area in which the HiSeqX Ten can shine because it’s faster and less expensive than many routine tests. Furthermore, whole-genome sequencing enables comprehensive genetic testing — from SNVs in known disease genes to large cytogenetic abnormalities — in a single assay. Ideally, both parents would undergo sequencing as well, to enable phasing of genetic variants and the identification of de novo mutations.
3. Cancer Genomes
Some of us have been preaching whole-genome sequencing for tumor genomes for a long time. The primary counter-argument has always been cost. The HiSeqX Ten essentially negates that issue. WGS for tumors has numerous advantages, including:
- More uniform coverage of coding exons than exome kits provide
- Genome-wide detection of SVs and copy number alterations, which are often pervasive (and clinically relevant) in tumor genomes
- Comprehensive somatic SNV discovery, which aids clonality and mutation rate analyses
In the past two years, our center has published two cases in which WGS revealed clinically-relevant genomic alterations that were missed by standard approaches. Uncovering a complex rearrangement of PML-RARA in a patient with PML-like disease, for example, led to a targeted therapy (ATRA) and complete remission for the patient.
Thus, whole-genome sequencing holds promise for individual cases, and also opens the door to the large-scale tumor sequencing projects (10,000 tumors or more) that would be required to fully characterize the significantly altered genes/pathways underlying a certain tumor type. One more advantage that may become especially important if the legal landscape of gene patents shifts. The Myriad Genetics patents on BRCA1/BRCA2 testing, even when enforceable, covered only the targeted sequencing of those genes in cancer patients. Whole-genome sequencing was never blocked by the patent; it was just too expensive.
4. Large-scale eQTL Discovery
One advantage of exome sequencing over genome sequencing is that it uncovers genetic variation which we are relatively well-equipped to interpret. It’s by no means simple, but we have the strong starting point of understanding that variants in coding sequences likely alter protein sequence and/or structure. In the other 98.5% of the genome, the analysis is much more challenging. Undoubtedly a significant proportion of important genetic variants lie in noncoding regions, where they exert a regulatory effect.
In other words, WGS will uncover variants that alter the expression or splicing of genes, as opposed to gene function. These differences will likely be quantitative, as opposed to binary. We have approaches in hand to search for expression quantitative trait loci (eQTLs) and splicing quantitative trait loci (sQTLs). Most of these compare gene expression data (microarray or RNA-seq) to high-density SNP array genotypes.
Large-scale genome sequencing coupled with RNA-seq, however, would provide the opportunity to comprehensively identify and characterize regulatory variation. It would be an ambitious undertaking to achieve the numbers required for statistical significance, but the payoff would be huge. In addition to improving our knowledge of and ability to identify regulatory variation, this would also help justify the cost, data storage, and effort required for whole-genome sequencing.