Recently I saw one of those presentations that seem common in the era of next-generation sequencing: the kind where they put up a list of genes that were variant or mutated or significant in a sequenced cohort. A glance at the list tells me that there are some true positives — one of the classic cancer genes is at the top — but there are other symbols I recognize, too: TTN, USH2A, MUC16…. we know them as the bad-apple genes, the ones that seem to crop up in any analysis.
With TTN and USH2A, it’s simply a gene size issue: these genes are so physically large that they tend to accumulate a lot of mutations in any cohort. And we see a lot of mucin genes because they’re recent gene copy events and give rise to many paralogous alignments, which in turn give rise to systematic false positive variant calls. For example, here’s an IGV screenshot of exome data for three samples at the MUC4 locus:
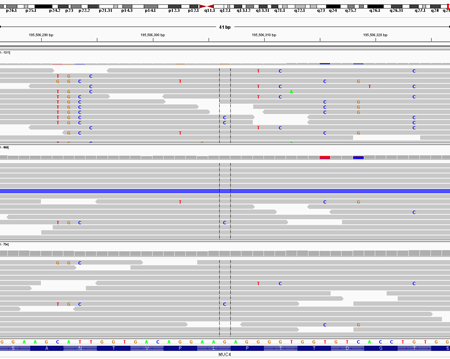
This represents what we call “paralogous alignment” and it’s one of the most frequent causes of false positive variant calls in NGS data. If you look at the pileup of these positions, the evidence for individual variants seems compelling because so many reads support the same variant at the same position. But these reads likely originate from another part of the genome, meaning that there’s probably not a real variant here.
There’s at least one notable exception to the expectation that a concentration of apparent variants in a small region is artifactual: the HLA locus on chromosome 6, which hosts the genes that encode human MHC proteins. In these regions there are many sequence variants, and most are probably real. However, the locus violates many of our assumptions about the human genome as a whole, which is why you often see a peak on chromosome 6 in Manhattan plot of genetic association signal.
False Positive Gene Lists
It gets to the point where analysts are building lists of these “usual suspect” genes as a ready, quick filter to remove them from candidate gene lists. Because treating TTN like it’s one of your top genes is kind of a rookie move. Incidentally, I learned that titin does have a role to play in humans, evidenced by the fact that targeted therapies against it cause heart failure. I recently made a list of genes that had novel heterozygous coding variants in multiple (unrelated) families with a rare Mendelian disorder. Then I took the first 3 characters of every gene name, and looked at how frequently that string occurred. Here were the top recurrences:
| Count | String | Description |
| 91 | LOC | LOC genes |
| 22 | ENS | Ensembl genes |
| 21 | FAM | FAM proteins |
| 15 | GOL | Golgi-like GOLGA8E |
| 13 | PRA | PRAMEF genes |
| 9 | NBP | Nuclear breakpoint family |
| 7 | POT | POTE ankyrin domain family |
| 6 | DEF | defensins |
| 5 | OR2 | Olfactory receptor |
| 5 | MUC | Mucins |
| 5 | KRT | Keratins |
| 4 | WAS | WAS protein family homolog |
| 4 | ANK | ankyrins |
| 3 | TRI | tri-partite motif containing |
| 3 | OR1 | Olfactory receptor |
| 3 | FRG | FSHD region gene |
The Danger of Filtering
Look at this list, and then think about how many papers you’ve read in which the “pathway analysis” of gene hits came up with olfactory or immune system pathways. Simply put, there’s a lot of protein sequence variation necessary for our sense of smell and our immune system, and underlying that is a great deal of genetic variation.
For rare Mendelian disorders, most of these are unlikely to harbor the causal variant. So it’s very tempting to simply filter them out and look at what’s left. There’s an inherent danger in that, however, since many of these are real genes that encode important proteins, and thus they could easily harbor variants that contribute to genetic disease.
Reducing False Positives from NGS
The next-gen sequencing community is not unaware of the challenges that false positives bring to bear on analysis of sequencing data. Many deterministic and/or statistical methods have been developed to help address them. For example, the significantly mutated gene (SMG) test in the MuSiC package considers gene size and local mutation rates in its assessment of whether a given gene is mutated more often than expected in a cancer dataset. And most variant calling pipelines employ advanced filtering strategies to remove systematic false positives that arise from common sequencing artifacts, like paralogous alignment.
The Need for a Better Reference
Many of the problems we face in NGS analysis will be mitigated by a better human genome reference assembly. This is because many parts of the human genome simply aren’t represented [accurately] in the reference, causing BWA and most other aligners to map them incorrectly. Worse, the aligners may not see anything wrong with such placements, and so they give the aligned reads a high mapping quality.
Of course, improving the human reference genome is no small request. Even with a better assembly in hand, releasing it, seeing it adopted by the NGS community, and updating all of our annotations of the human genome to its coordinate system requires a huge amount of work and time. In the end, however, it will give us better analysis results, and improve our ability to understand the genetic basis of human disease.
Well, you’re running the right mutation callers, and you don’t have many options for detecting “driver” mutations with a single tumor. Basically, you’re hoping to get lucky, and see a mutation in the tumor and clones that’s in a known or likely cancer gene. Or, a gene that you could follow up with some functional assays.
TTN is not regarded as a cancer gene by most of the analysts I know. It simply acquires many coding mutations because it’s so large. The mucins also turn up a lot, but I think that’s because there are so many genes that are similar to one another that you get spurious mutation calls from paralogous alignments.
For these reasons, we’ve ignored genes like TTN and the mucins in the past when they turned up in SMG lists. Newer evolutions of the significantly mutated gene algorithms do a better job of accounting for gene size and mutation rate, causing the genes to drop off the lists altogether. As they probably should. Of course, there are no absolutes — some mucin genes probably do harbor cancer-relevant mutations. I’ve even heard from colleagues that if you knock out TTN in mice, it’s embryonic lethal.
vd4mmind I don’t have one, but I think our colleagues at the Broad have put something like that together. And the 1,000 Genomes Project made some kind of a blacklist (of variants that were false positives); that might be useful, too.