About 1 in 8 women in the U.S. will develop breast cancer at some point in her lifetime. In 2013, an estimated 40,000 breast cancer deaths will occur in the U.S. and 300,000 women will be diagnosed with invasive or in situ malignancies.
It’s not only one of the most common cancers, but it’s also one that can run in families. About 5-10% of breast cancers are associated with highly penetrant germline (inherited) susceptibility alleles, a figure supported by the TCGA Breast Cancer study.
The best known susceptibility genes are BRCA1 and BRCA2, whose discovery more than 20 years ago launched a sustained effort to identify new genes explaining the missing heritability in the disease. Today, known susceptibility genes such as BRCA1/2, TP53, PTEN, STK11, PALB2, and ATM account for about 35% of familial breast cancer cases.
Notably, as reported today by GenomeWeb, the genetic test for BRCA1/BRCA2 in women with atypical breast cancer diagnoses (young age or family history) will be covered by the Affordable Care Act with no copay. I wonder what the outspoken haters-of-Obamacare will say to that.
Back to breast cancer susceptibility. Right now, two-thirds of inherited risk comes from as-yet-unidentified genes. It is reasonable to assume that the most common and highly penetrant susceptibility genes have already been discovered. Now we’re looking for rare variants and/or low-penetrance alleles, which will be more challenging to identify because thousands of samples are required. Are such cohorts available? Yes. But the cost to sequence them all, despite advances in next-generation instruments, remains prohibitive.
Multi-Step Studies of Rare Variants in Disease
A recent study in PLoS ONE by Gracia-Aznarez et al demonstrates that we can uncover rare susceptibility variants using a multi-step strategy:
- Discovery sequencing (exome or whole-genome) in a small number of families.
- Prioritization of candidate genes/variants using segregation (within a family) and control cohorts
- Targeted evaluation of candidates in thousands of cases and controls
This type of strategy (discovery, prioritization, extension) has already been exploited for studies of tumor genomes. While obviously not as powerful as a whole-genome sequencing study on everyone right off the bat, it’s economically feasible. And, statistically, this study design does two clever things. First, it provides (in step 3) the thousands of samples required to properly evaluate rare variants. Second, by specifically targeting certain variants or genes, it dramatically reduces the penalty of multiple testing.
Rare Variants in Familial Breast Cancer
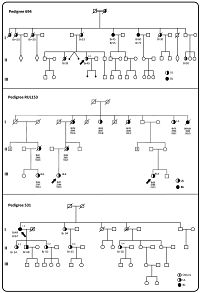
In the present study, the authors performed exome sequencing on eleven cases from 7 families that:
- Had at least 6 women diagnosed with breast cancer before the age of 60
- Lacked known genetic variants in BRCA1/BRCA2
- Had no history of ovarian cancer
These criteria enrich for families likely to have new susceptibility genes of some penetrance. The ovarian cancer requirement was a bit surprising, since BRCA1/2 mutations also predispose to that malignancy. Also, the index cases from the extension cohort comprised some families with breast and ovarian cancer.
Seven controls were also sequenced; these were HapMap cell lines (NA11881, NA12144, NA12750, NA12761, NA12763, NA12813, and NA12892). It’s kind of a waste of money for everyone to keep sequencing HapMap samples as controls, since they’re bound to be exome (if not whole-genome) sequencing data for all of these samples. And we don’t know if they’re true controls, i.e., individuals who won’t get breast cancer. But I digress.
Filtering and Genetic Association
After exome capture (Agilent) and sequencing (Illumina GAIIx), the authors mapped reads to the hg18 reference sequence (Novoalign) and called SNPs and indels (regrettably using SAMtools and not VarScan). They next employed a series of filters to nominate candidates for further analysis, prioritizing variants that were:
- Present in both members of the family (if two were sequenced)
- Absent from the 7 HapMap controls and dbSNP 130
- Predicted to alter protein sequence (nonsynonymous coding)
- Affected a gene with a potential role in cancer (not sure how this was determined)
It’s hard not to focus on the weak points of the analysis here — the small number of controls, the use of the NCBI36/hg18 reference (now 7 years old), the mysterious “score and gene function” filter that helped reduce the number of candidate variants from 60,000 to 81. I don’t want to dwell on that, because I think the general study design, if not the detailed methodology, makes for a good example.
Rare Variants Associated with Breast Cancer
Candidate variants of interest were validated by 3730 and segregation studies, checked in a population of Spanish controls, and then evaluated by TaqMan in ~2000 cases and ~2000 controls. At the end of it, the authors had a known moderate susceptibility variant (CHEK2 1100delC) and a “catalogue” of 11 rare variants in other genes:
- FANCM, a fanconi anemia gene
- WNT8A, a member of wingless-type MMTV integration site family
- MAPKAP1, a protein associated with MAP kinase
- TNFSF8, from the tumor necrosis factor ligand superfamily
- PTPRF, a protein tyrosine phosphatase receptor
- UBA3, an ubiquitin-like modifier activating enzyme
- AXIN1, a component of the beta-catenin destruction complex
- TIMP3, a TIMP metallopeptidase inhibitor
- SLBP, a stem-loop binding protein
- CNTROB, a centrosomal BRCA2-interacting protein
- S1PR3, a sphingosine-1-phosphate receptor
Most of these sound interesting as potential cancer susceptibility genes, while some might be hard to tell a story about right now. Undoubtedly, as more ambitious efforts are undertaken and published, we’ll continue to round out the catalogue of genetic variation underlying breast cancer susceptibility.
References
Gracia-Aznarez FJ, Fernandez V, Pita G, Peterlongo P, Dominguez O, de la Hoya M, Duran M, Osorio A, Moreno L, Gonzalez-Neira A, Rosa-Rosa JM, Sinilnikova O, Mazoyer S, Hopper J, Lazaro C, Southey M, Odefrey F, Manoukian S, Catucci I, Caldes T, Lynch HT, Hilbers FS, van Asperen CJ, Vasen HF, Goldgar D, Radice P, Devilee P, & Benitez J (2013). Whole Exome Sequencing Suggests Much of Non-BRCA1/BRCA2 Familial Breast Cancer Is Due to Moderate and Low Penetrance Susceptibility Alleles. PloS one, 8 (2) PMID: 23409019