Next-generation sequencing technologies have transformed the way we study rare genetic disorders. Exome sequencing for Mendelian disease is the poster child for success in this arena. However, despite the frenzied pace of disease gene discovery and the growth of public sequence databases, the diagnostic/success rates for exome sequencing have remained somewhat constant over the past several years. Depending on the type of disorder, available samples, mode of inheritance, and other factors, one can expect that ~35-50% of clinical cases may be solved by exome sequencing.
On the research side, the success rates for rare disease studies have often gone down, for the simple reason that as we improve the catalogue of disease-causing genes/mutations, genetic tests become increasingly effective. In other words, easy-to-solve cases never make it to a research study. The cases that do make it through, while valuable for gene discovery, will almost certainly have a low success rate.
Knowing this intellectually does not make it any less frustrating when you can’t solve a rare disease patient or family where there’s clearly something to find. When an exome is negative, what recourse do we have? Whole-genome sequencing is often what researchers consider pursuing next — in theory, it enables the detection of variants that are not detectable by exome sequencing (e.g. in the 5-10% of coding bases that exome sequencing does not sufficiently cover, or structural variants with noncoding breakpoints). Yet the “rescue rate” of WGS for negative exome cases has been disappointing.
Transcriptome sequencing offers a possible alternative to further investigate negative exome cases. For certain categories of variants (e.g. nonsense, frameshift, and splice site mutations), RNA-seq can provide a molecular confirmation of the predicted pathogenic effect. This is especially helpful in the gray area of splice-region variants, the effect of which is difficult to assess from genomic information alone.
A recent paper from the MacArthur lab demonstrates the utility of RNA-seq in patients with genetically undiagnosed rare muscular disorders. They applied RNA-seq to muscle tissue from 50 unsolved cases. Four of these had a splice region variant of uncertain significance. Another 12 had a strong candidate gene based on clinical presentation and/or the presence of a single pathogenic variant in a known recessive gene. The majority (34) had undergone exome/genome sequencing but yielded no candidates.
Pathogenic Extended Splice Variants (2 cases)
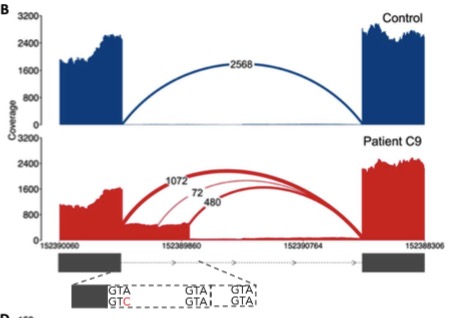
Exon extension caused by a donor +3 A>C extended splice site variant (B. Cummings et al, Sci Transl Med 2017)
First, the straightforward analysis. In the four cases with candidate variants in the extended splice region (i.e. not the canonical splice site, but close to it), RNA-seq confirmed pathogenicity in 2 of them. This is a small sample size, so all it really does is highlight the difficulty of predicting variant effects outside of the canonical splice site.
Unrecognized/Undetected Splice Variants (4 cases)
Interestingly, RNA-seq enabled the diagnosis of a three cases in which a splicing variant was not suspected:
- 1 with a splice-region variant was pathogenic but had been missed by exome sequencing
- 1 with a missense variant that alters splicing
- 2 synonymous variants that alter splicing
The missense variant is interesting: it creates a stronger splicing motif than the wild-type sequence, leading to an exonic splice gain:
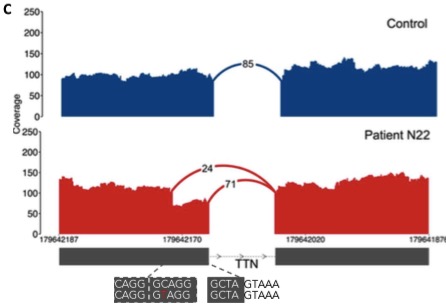
Exonic splice gain caused by a C>T donor splice site–creating variant in patient N22
Deep Intronic Variants that Alter Splicing
The real power of RNA-seq in cases like these is to identify novel splicing events that are not due to obvious splice site/region variants. In seven patients, the authors of this study identified aberrant splicing caused by deep intronic variants in known disease genes (DMD and COL6A1).
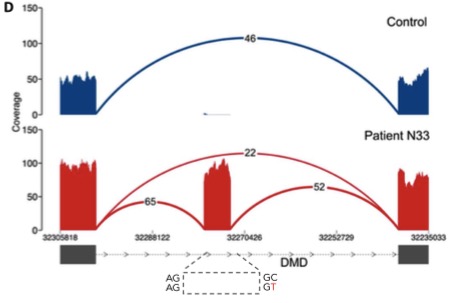
Intronic splice gain in patient N33 caused by a C>T donor splice site–creating deep intronic variant.
RNA-seq is really the only approach that can identify an event like this. Exome sequencing doesn’t cover this region. Whole-genome sequencing does, but undoubtedly will return dozens of intronic variants, most of which have no effect on splicing.
Rescue Rate for RNA-Seq
Overall, transcriptome sequencing enabled the diagnosis of 17 patients, or 34%. This rate is skewed a bit high by cases with candidate splice region variants (2/4 solved) and cases in which there was already a strong candidate gene (66%). Among cases without such strong leads, the rescue rate was 21%. This is a not-inconsiderable improvement to the overall diagnostic rate, and a strong argument that RNA-seq should be considered as a possible next step when exome sequencing alone does not achieve a diagnosis.
Importance of the Right Tissue
It should be noted that muscular disorders were well-suited to a transcriptome approach, because the relevant tissue is both accessible and often already banked due to the fact that muscle biopsy is a routine (if painful) diagnostic procedure. For other types of rare disorders, one may not have this luxury. In retinitis pigmentosa, for example, the relevant tissue is the retina. Good luck getting any RP patients to volunteer any of their diminishing retinal cells. Developmental timing is also a consideration: for some disorders, like birth defects, the genetic aberration affects a certain phase of development that is no longer ongoing when the patient is in the hospital.
You’ll note that the figures shared in this post also included, in blue, the splice pattern observed in normal individuals. These were gleaned from a subset of the skeletal muscle samples sequenced by the GTex consortium that were carefully matched to the patient specimens. A tissue-matched normal is incredibly powerful for analyses such as this one, and reinforces the need for more large-scale efforts to put transcriptome data in the public domain.