As you probably know, I’m a fan of exome sequencing, particularly for studies of rare inherited disorders. While not a perfect (or comprehensive) assay, exome sequencing offers an efficient screen of the regions most likely to harbor disease-causing mutations. Ironically, another reason people like exome sequencing is because of the limited scope: it essentially doesn’t interrogate the regions (noncoding) and variant types (SVs) that are more difficult to interpret.
Well, the party’s over.
Several large scale whole-genome sequencing studies of human disease are hurtling forward in the U.S., the U.K., and other countries. We’ve put it off as long as we can, but now we’re faced with the daunting task of identifying and interpreting biologically-relevant variants outside of protein-coding exons.
There’s good reason to do so, by the way, especially studies of common disease in which regulatory activity is likely to play an important role. Many, if not most of the genomic loci associated with human traits lie in noncoding regions. According to datasets generated by projects like ENCODE and FANTOM5, noncoding regions also exhibit an astonishing amount of biochemical activity suggestive of diverse functions.
Sometimes with a daunting analysis task, it’s hard to know where to start. Fortunately, there’s a nice paper in the upcoming issue of AJHG that provides some practical guidance. Alanna C. Morrison et al present a series of integrated steps for whole-genome analysis and apply them to study 10 heart- and blood-related traits in 1,860 African Americans.
The authors perform aggregation tests (sometimes called burden tests) to test sets of rare variants for association. Such tests require that one define a unit by which to group variants. With exome sequencing, this was commonly done on a per-gene or per-exon basis. In this study, with WGS available, the authors aggregated variants:
- In a sliding 4-kb window across the entire genome
- In the first introns of protein-coding genes, which are known to harbor regulatory elements
- In pre-defined regulatory domain motifs (promoters, enhancers, and UTRs) near genes.
This is, in modern-day terms, a modestly powered study of complex quantitative traits. Even so, the authors found several significant associations, some near known loci for these traits, and others in potentially novel regions. The results of their complementary methods are illustrated in Figure 3:
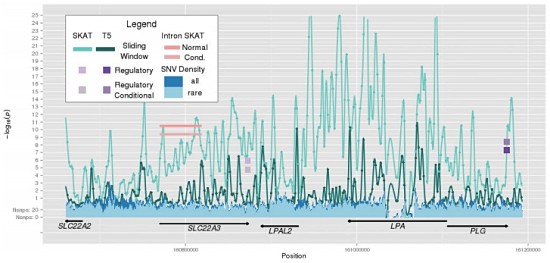
Morrison et al, Am. J Hum Genet, 2017
You’re looking at the association of Lp(a) levels in the well-known LPA gene locus. The sliding window approaches nicely captured association over the LPA gene region, but also in other nearby regions, some of which were also supported by the first-intron or regulatory-motif analyses. It’s a complicated picture, to be sure, but it suggests some specific areas in which noncoding variants are exerting a cis-regulatory effect on the LPA gene. Very cool stuff.
What’s especially nice about this paper is that it provides relatively straightforward methodology for tackling the daunting analysis task I talked about above:
- Obtain WGS data for a well-phenotyped cohort
- Define some common-sense strategies for aggregating (grouping) rare variants
- Apply aggregation tests on a genome-wide basis
- Replicate significant findings in an independent cohort (in this case, about 2,000 European-Americans)
This study demonstrated both the feasibility and the justification for interrogating noncoding regions for association with medically important traits. Imagine how much we’ll be able to discover as we get our hands on massive WGS cohorts, and extend these principles to other regions and regulatory motifs of the genome.