De novo mutations — sequence variants that are present in a child but absent from both parents — are an important source of human genetic variation. I think it’s reasonable to say that most of the 3-4 million variants in any individual’s genome arose, once upon a time, as de novo mutations in his or her ancestors. In the past few years, whole-genome sequencing (WGS) studies performed in families (especially parent-child trios) have offered some revelations about de novo mutations and their role in human disease, notably that:
- The de novo mutation rate for humans is ~1.2e-08 per generation which works out to around 38 mutations genome-wide per offspring
- 95% of the global mutation rate is explained by paternal age (each year adding 1-2 mutations)
- As much as 2/3 of genetic diagnoses from clinical sequencing efforts are de novo mutations.
A recent study in Nature Genetics provides the largest survey of de novo mutations to date. Laurent Francioli et al identified de novo mutations in 250 Dutch families that were sequenced to ~13x coverage as part of the Genome of the Netherlands (GoNL) project. Their findings confirm much of the observations from previous smaller studies, and offer some new insights into the patterns of de novo mutations throughout the human genome.
Identification of de novo Mutations
To make any global observations about de novo mutations, one generally needs unbiased whole-genome sequencing data for an individual and both parents. Even with those in hand, accurate identification of de novo mutations is challenging because they’re so exquisitely rare. Since the sequencing coverage in this study is a little bit light (13x, whereas most studies shoot for ~30x), I had some initial concerns about whether or not the mutation calls might hold up under scrutiny.
Delving into the online methods, I learned that the samples underwent Illumina paired-end sequencing (2x91bp, insert size 500bp). Alignment and variant calling followed GATK best practices (v2), and the mutations were called with the trio-aware GATK PhaseByTransmission. Next, the authors used a machine learning classifier trained on 592 true positive and 1,630 false positive de novo calls that had been validated experimentally. The net result was 11,020 high-confidence mutations in the 269 children, with an estimated a 92.2% accuracy.
The numbers are about right: if 92.2% of the calls are real, that’s 10,160 true mutations, or ~37.7 mutations per child. That’s very close to the estimated ~38 per genome. In other words, without experimentally validating all 11,000 mutations (an expensive and laborious task), this is as good as it gets.
Parent-of-Origin and Replication Timing
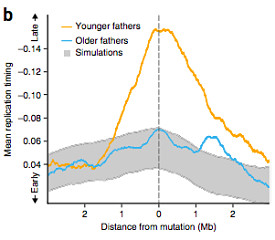
Credit: Francioli et al, Nature 2015
The authors first examined whether the location of the observed mutations was correlated with any epigenetic variables. There was no significant correlation for most of the variables examined (chromatin accessibility, histone modifications, and recombination rate). With a linear regression model, they noted a significant association between replication timing and paternal age: mutations in the offspring of younger fathers (<28 years old) were strongly enriched in late-replicating regions, whereas mutations in offspring of older fathers were not.
To dig deeper, the authors looked at 2,621 mutations that could be unambiguously assigned to maternal or paternal origin. The method for this isn’t documented in the online methods, but presumably they looked for instances in which a mutation was in the same read or read pair as a variant unique to one parent. Notably, 1,991 of those origin-inferred mutations (76%) came from the father. After controlling for the number imbalance, the replication-timing-with-parent-age correlation was significant only for mutations of paternal origin.
This makes a certain kind of sense, since the stem cells in the paternal germ line undergo continuous cell division throughout a man’s life, whereas a woman is born with all of the eggs she’ll ever have.
The correlation between paternal age and replication timing is important from a reproductive health perspective, because late-replicating regions have lower gene density and expression levels than early ones. Since the mutations in offspring of younger fathers tend to occur in these regions, they’re less likely to have a functional impact. In support of this idea, on average, the offspring born to 40-year-old fathers had twice as many genic mutations as offspring born to 20-year-old fathers.
In other words, mutations in the offspring of older fathers are not only more numerous, but also more likely to have functional consequences.
Mutations in Functional Regions
Notably, the de novo mutation rates in this study were higher in exonic regions regardless of the paternal age. Overall, 1.22% of mutations were exonic, an enrichment of 28.7% over simulated models of random mutation distribution. Mutations were also enriched in DNase I hypersensitive sites (DHSs), which represent likely regulatory regions. The source of this “functional enrichment” likely has to do with sequence context: mutations often occur at CpG dinucleotides, which are themselves more prevalent in exons and DHSs.
Recent studies of somatic mutations in tumor cells revealed a fascinating phenomenon: a reduction in the mutation rate of highly transcribed regions, likely attributed to the fidelity conferred by transcription-coupled DNA repair mechanisms. In the current study of de novo mutations, however, the mutation rate in transcribed regions and DHSs did not appear to be reduced.
The implication here might be that transcription-coupled repair has less of an impact on de novo mutations, though the authors note that their study was only powered to detect a substantial difference (>17%) in mutation rate. That’s understandable, because while the individuals examined here harbored ~40 mutations genome-wide, a tumor specimen might have tens of thousands of somatic mutations (i.e. much better power to detect subtle differences in mutation rate).
Clustered de novo Mutations
One of the most interesting observations in this study was a clustering effect of de novo mutations. If all things were random, given the size of the genome (3.2 billion base pairs) and the number of mutations per individual (~40), we expect them to be pretty far apart. As in, one every 80 million base pairs.
Instead, the authors observed 78 instances in which there were “clusters” of 2-3 mutations within a 20kb window in the same individual. The 161 mutations involved showed no significant differences from the non-clustered mutations with regard to recombination rate (p=0.52) or replication timing (p=0.059), though I should point out that the latter might be approaching an interesting p-value.
Interestingly, however, the clustered mutations exhibited an unusual mutational spectrum, with a strong enrichment for C->G transversions compared to non-clustered mutations (p=1.8e-13).
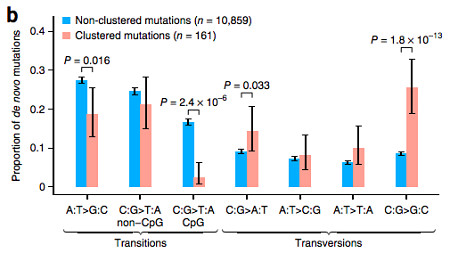
Francioli et al, Nature 2015
Based on the nucleotide context, the authors suggest that a new mutational mechanism may be at work involving cytosine deamination of single-stranded DNA (presumably during replication). I don’t have strong enough chemistry to understand the proposed mechanism, but agree that this unusual pattern merits some more investigation.
References
Francioli LC, Polak PP, Koren A, Menelaou A, Chun S, Renkens I, Genome of the Netherlands Consortium, van Duijn CM, Swertz M, Wijmenga C, van Ommen G, Slagboom PE, Boomsma DI, Ye K, Guryev V, Arndt PF, Kloosterman WP, de Bakker PI, & Sunyaev SR (2015). Genome-wide patterns and properties of de novo mutations in humans. Nature genetics, 47 (7), 822-6 PMID: 25985141