The past few decades have seen rapid advances in our knowledge of genetic diseases, which affect an estimated 25 million Americans. These advances can be quantified in things like the growth of dbSNP (now contains about 90 million validated genetic variants) and the number of Mendelian disorders understood at the genetic level (over 5,000).
Some of the factors that have contributed to this progress include:
- Big science. Ambitious, grant-supported, international efforts like the Human Genome Project, the HapMap Project, and the Cancer Genome Atlas yielded the public resources that form the foundation of modern human genetics research. Thank you, taxpayers.
- Technology development. Revolutionary advances in genome interrogation technologies (high density SNP arrays, whole-genome sequencing, etc.) have made large-scale genetic studies feasible, both technically and financially.
- Study participants. It’s important to remember that most (if not all) of human genetics studies could not have happened without the patients and families who volunteered their samples, often with the knowledge that they’d get nothing in return.
The Unsolved Problem of Inherited Disease
Few areas have benefited as much from these advances as the study of rare genetic diseases. Exome sequencing has enabled the rapid genetic diagnosis of many patients, and the discovery of hundreds of new Mendelian disease genes. Yet even well-powered Mendelian disease studies can fail for a variety of reasons. There’s also a considerable gray area between success and failure: the implication of an unknown gene, or one that has never been associated with disease.
One particular challenge is that Mendelian diseases are rare by definition, and the variants definitively shown to cause them are rarer still. As a result, many variants detected in clinical sequencing project end up with the label variant of unknown significance, or VUS. Even when given a classification, some variants are interpreted differently by different clinical laboratories.
As discussed in a report at the New England Journal of Medicine this week, another thing that has hampered our ability to discover and annotate clinically-relevant genetic variation is the “silo effect” — in which research groups (both commercial and academic) maintain private databases of clinical sequencing results. A great example of this is Myriad Genetics, a company that’s probably sitting on the largest database of BRCA1/2 mutations in the world.
The problem, of course, is that not all of the clinical datasets for a given disease or gene ends up in the same silo. Thus, researchers in group A might have a promising new disease gene that researchers in group B have also identified in a different family kindred. If those datasets were shared, rather than kept isolated, these groups could cross-validate with one another and the research community as a whole would benefit.
Data Sharing in ClinVar
The NIH’s Clinical Genome Resource program (ClinGen) hopes to address some of these issues by developing community resources to understand our understanding of genomic variation and improve its use in clinical care. The cornerstone of this effort is ClinVar, a database of variants annotated with clinical data.
ClinVar Contributors
Over 300 different submitters have contributed to ClinVar thus far. Those submitters comprise research groups, clinical laboratories, locus-specific databases, and aggregate databases (like OMIM). Here’s a plot of the variants submitted for some of the major (or interesting) contributors:
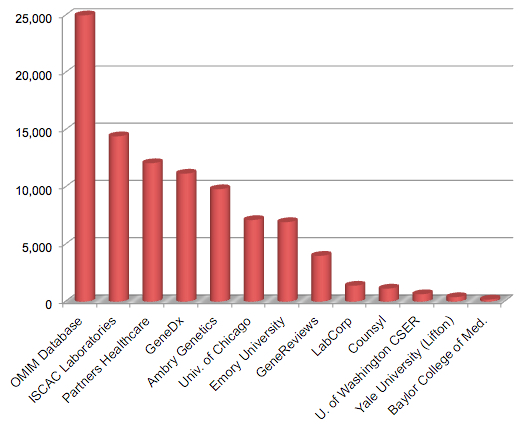
Selected ClinVar Submitters (adapted from Table 2, Rehm et al, NEJM 2015)
The largest submitter by far is OMIM, which has contributed over 25,000 variants to ClinVar. It’s encouraging to see two of the leading genetic testing providers (GeneDx and Ambry Genetics) making substantial contributions. Among academic centers, the University of Chicago and Emory University are the clear leaders.
As of May 2015, ClinVar contained 172,055 variant submissions across 22,864 genes. More than 118,000 unique variants have clinical annotations, though 21% of those are “variant of unknown significance.” Nevertheless, this rapidly-growing resource illustrates the power of sharing clinical variant annotations in a centralized manner.
Discordant Clinical Annotations
Notably, 12,895 variants have clinical annotations (pathogenic, unknown, or benign) from at least two different laboratories and 17% of the time, those annotations did not agree. For example, at least 220 of the “pathogenic” variants pulled in from OMIM (the largest contributing database) are classified by clinical laboratories as either benign or unknown significance.
It is clear that the guidelines for variant interpretation differ between laboratories, and need to be standardized. Even so, adopting standards and making the effort to share clinical variant findings and annotations (along with the relevant phenotype data) is critical to the success of rare disease research. ClinVar seems to be taking us in the right direction.
References
Rehm HL, Berg JS, Brooks LD, Bustamante CD, Evans JP, Landrum MJ, Ledbetter DH, Maglott DR, Martin CL, Nussbaum RL, Plon SE, Ramos EM, Sherry ST, Watson MS, & ClinGen (2015). ClinGen – The Clinical Genome Resource. The New England journal of medicine PMID: 26014595