Although the cost of sequencing continues to fall precipitously (cue the NIH sequencing-versus-Moore’s-Law figure), it’s still expensive relative to high-throughput genotyping. Whole-genome sequencing on the X Ten costs around $2500 per sample by the time you account for basic analysis and data storage. This means that a well-powered genetic association study for complex disease (10,000 samples) would cost over $20 million just for data generation. The same cohort genotyped on a high-density SNP array might only cost about $1 million. Undoubtedly, that’s why most large scale genome-wide association studies to date (>50,000 samples) have relied primarily on SNP array data.
There is a growing body of evidence, however, that rare variants (especially ones not present on SNP arrays) might confer a significant proportion of the genetic risk for complex disease. In age-related macular degeneration (AMD), for example, sequencing studies of moderate size (~5,000 samples) were able to identify rare coding variants in C3 and CFH associated with risk of disease. An important advantage of a sequencing approach is the ability to perform aggregation tests of private and rare coding variants (e.g. with the sequence kernel association test, SKAT) to boost the power to detect association.
A recent paper in Nature Genetics illustrates the feasibility of this approach for sequencing studies of complex disease. Stacy Steinberg and colleagues from deCODE Genetics conducted a search for rare functional variants in the known risk loci for Alzheimer’s disease (AD) using a unique resource: whole-genome sequences of 2,636 Icelanders imputed into 104,220 long-range phased individuals and their relatives.
So here we have a rare variant association study (RVAS) that employs several strategies for an efficient design:
- Studying an isolated population (Iceland), whose genetic structure enabled accurate genotype imputation of a large sample set (>100k individuals) with sequencing data for just 2,500.
- Analyzing missense variants with SKAT, which aggregates rare variants (i.e. collapses them at the level of the gene) to boost power for association but allows for multiple directions of effect.
- Examining only regions known to be associated with AD — which seem likely to harbor [rare] functional variants — to reduce the multiple testing penalty.
Targeted Association Studies
There are, of course, disadvantages to limiting the scope of association testing to known regions. Obviously you won’t be discovering any new associations, especially ones that sequencing (but not genotyping) might be able to uncover. Even so, you’re stacking the deck in your favor because the known GWAS loci almost certainly harbor some functional variation that hasn’t yet been fully interrogated.
Sometimes, sequencing will only serve to replicate the common variant association signal (i.e. not find anything new). Yet these targeted approaches might help narrow the boundaries of the associated region — which could encompass dozens or hundreds of genes — or, even better, identify disruptive variants whose LD with the lead SNP makes them good candidates for causal variants. Thirdly, one might uncover secondary independent association signals in GWAS loci, implicating that there are multiple haplotypes that influence disease risk.
Variant Annotation and Aggregation
As anyone who has done aggregation/burden testing in association studies can tell you, the analysis choices can have a significant impact on results. The annotation tool/source, MAF threshold, and variant mask (definition of what’s deleterious and should be included) can introduce a lot of variability. In this case, the authors tried two variant masks:
- Loss of function variants: nonsense, frameshift or canonical splice site variants. These are usually quite rare, and so the authors collapsed them to a single “meta variant” at the level of the gene.
- Missense variants: nonsynonymous variants or splice region variants. This latter one is an interesting choice, and not necessarily one I’d have thought to make at the discovery stage.
The burden tests included only variants with MAF<1% and information (call rate) >0.80. The authors tested about 80 genes across the 17 loci, and the top-scoring hit was ABCA7 (p=0.00020).
Splice Region Variation in ABCA7
ABCA7 encodes ATP-binding cassette transporter A7, a member of ABC transporters that move lipids across membranes. The SKAT result was primarily driven by a single variant, c.5570+5G>C. Without it, the test had a p-value of 0.46. If you’re familiar with the notation, then you know that c.5570+5 indicates a noncoding variant 5 bases into an intron. We call this the “splice region” and, unlike the canonical splice site (+/- 2bp) it’s not clear that variants here affect splicing.
But the authors had another NGS tool to look at this: RNA-seq. When they looked at the transcript sequences of c.5570+5G>C carriers, they included a retained intron that eventually included a stop codon.
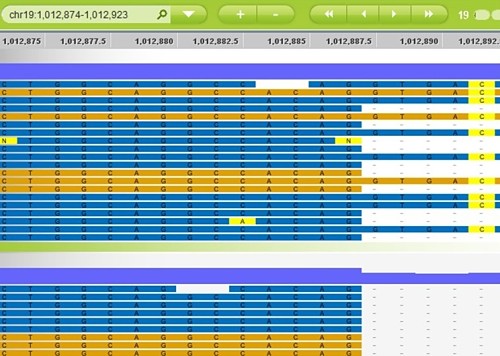
Intron retention in carriers (Steinberg et al, Nat. Gen. 2015, Fig S1)
The image here is from Supplemental Figure 1 (the main text had no figures) and shows the intron retention in c.5570+5G>C carriers. Side note: according to the legend, the coordinates are on NCBI build 36, which practically a crime. But moving on, the RNA-seq results justified including the variant in the loss-of-function test (mask #1), which then yielded a p-value of 5.3e-10 with odds ratio of 1.97.
Follow-up and Replication of Association
With a possible causal variant in hand, the authors next examined the long-range haplotypes to see if this variant was on the same background as rs4147929, the common variant previously associated with AD by GWAS. It was never on the same allele, which is a fascinating result; the common variant signal and this rare variant association appear to be independent. It’s possible, therefore, that the mechanisms are different as well.
To replicate the association, the authors genotyped ABCA7 loss-of-function variants in study groups from Europe and the United States, finding a p-value of 0.0056 with OR of 1.73. When combined with the Icelandic data by meta-analysis, the OR was 2.03 and the p-value 6.8e-15.
What’s Next for AD and Common Disease
ABCA7 certainly merits future studies, both in the genetics realm and in the laboratory for functional evaluation. It’s strongly expressed in the brain, where it promotes the efflux of phospholipids and cholesterol to apoA-I and apoE. But the ortholog of ABCA7 in C. elegans and results from mouse models suggest that regulation of phagocytosis might be the primary function of the gene. The authors tested for correlation between variants in ABCA7 and two disease-associated alleles (in APOE and TREM2), but found none. Thus, the mechanism by which ABCA7 loss-of-function confers susceptibility to AD will need further investigation.
Still, it’s a promising start to detangling the etiology of a complex human disease, and a demonstration of the power of genome sequencing to uncover promising new leads.
References
Steinberg S, Stefansson H, Jonsson T, Johannsdottir H, Ingason A, Helgason H, Sulem P, Magnusson OT, Gudjonsson SA, Unnsteinsdottir U, Kong A, Helisalmi S, Soininen H, Lah JJ, DemGene, Aarsland D, Fladby T, Ulstein ID, Djurovic S, Sando SB, White LR, Knudsen GP, Westlye LT, Selbæk G, Giegling I, Hampel H, Hiltunen M, Levey AI, Andreassen OA, Rujescu D, Jonsson PV, Bjornsson S, Snaedal J, & Stefansson K (2015). Loss-of-function variants in ABCA7 confer risk of Alzheimer’s disease. Nature genetics PMID: 25807283