In the last decade, genome-wide association studies (GWAS) enabled by cheap, high-throughput SNP genotyping have identified thousands of loci that influence disease susceptibility, quantitative traits, and other complex phenotypes. The genetic markers on high-density SNP arrays are carefully chosen to capture (or “tag”) most common haplotypes in human populations. Common SNPs tend to be more informative in this regard, and most of these fall outside the exons of protein-coding genes.
Credit: Leslie et al, AJHG 2015
This efficiency is both the strength and the weakness of SNP arrays: they are well-suited to represent variation across the human genome, but they’re unlikely to be causal variants themselves. In essence, the loci uncovered by GWAS are signposts that tell us where to look for functional variation that influences a trait of interest.
Following up GWAS hits — with sequencing and functional validation — will ultimately be required to understand the mechanism of disease. A paper online at The American Journal of Human Genetics offers an informative example of how that plays out.
Cleft Lip/Palate as a Complex Trait
Non-syndromic cleft lip with or without cleft palate (NSCL/P) affects about 1 in 700 live births, and represents a global health problem (particularly in the developing world). Multiple genetic and environmental risk factors give rise to a complex etiology for this trait. One candidate gene (IRF6) was known to harbor common variants associated with NSCL/P, and large-scale GWAS efforts have yielded 12 additional loci reproducibly associated with it.
To further investigate the genetic architecture of this phenotype, a group of researchers from several institutions (including the Genome Institute at WashU) sequenced those 13 regions in over 4,000 individuals. At the study design stage, the researchers made two key decisions that undoubtedly contributed to their success:
- A case-parent trio design. Most of the samples chosen came in the form of an affected child and two unaffected parents. This structure makes it possible to examine not just the presence or absence of alleles, but whether or not they’re transmitted to the affected child. It also permitted a search for de novo mutations that might contribute to susceptibility.
- A wide target region for each locus, including both coding and non-coding regions. The latter type are increasingly important as we delve into complex traits in which a significant fraction (if not a majority) of causal variants will be regulatory rather than coding in nature.
De novo Mutations
Just as one can’t truly identify somatic mutations in a tumor tissue without a matched normal, it’s nearly impossible to distinguish de novo mutations in a patient without sequencing both of his or her parents. This is the only way, people. Filtering dbSNPs is not going to get you there.
The thing about de novo mutations is that they’re exquisitely rare — according to estimates of the de novo mutation rate, any given individual should have around 34 mutations genome-wide. Since the target space for this study represented 0.19% of the genome, that’s a long shot. Then again, we’re talking about a lot of trios, and they’re selected for a trait that’s been linked to this target space.
My back-of-the-envelope calculations based on the amount of target space (6.3 Mbp), the de novo mutation rate (1e-08), and the number of trios sequenced here (1,409) suggest that we’d expect ~89 de novo mutations. The authors came up with 123, which is a little high. We do have an enriched population, but calling de novos is very likely to yield some false positives.
They were able to design assays for 82 mutations and confirmed 66 (80%) by Sanger sequencing. That’s a good validation rate, and it suggests that about 98 of the predicted mutations would hold up (pretty close to my estimate).
Only 3 of 66 confirmed de novo mutations (3.6%) altered protein sequence. The majority (95%) were noncoding, though 11 of these mapped to a predicted regulatory element.
Common Variant Associations
To identify common functional variants, the authors used an allelic transmission disequilibrium test (TDT), which determines if an allele is transmitted more (or less) than we’d expect by chance. All but one of the GWAS regions (PAX7) showed evidence of association with p-values less than 10-5. In general, the results supported the GWAS findings: the variants yielding the lowest p-value were either the lead GWAS SNP or were in perfect LD with the lead GWAS SNP.
A conditional analysis revealed only one locus (ARHGAP29) with evidence for secondary independent signals, suggesting more than one common functional variant.
Rare Variant Associations
The common variants explained only a fraction of the heritability for disease. Yet these GWAS regions were also logical candidates for rare variation that contributes to disease. The challenge with rare variants is that one needs thousands of samples to even see them, much less establish statistically significant association.
To address this, we often collapse individual rare variants to the gene, regulatory element, or genomic interval in which they occur to get the power up. These so-called burden tests can boost the power for detection, but that’s dependent on one’s ability to predict which variants are truly functional. In this study, neither gene-based or regulatory element-based burden tests yielded significant associations.
A ScanTrio analysis of genomic intervals — after experimenting with different window sizes and overlaps — yielded signals for 2 of the 13 regions (NOG and NTN1).
Functional Validation
The challenge of genetic association studies — especially ones for complex phenotypes — is confirming statistical evidence of association with a functional assay. Sequencing and genotyping methods continually become faster and cheaper. With functional validation, the only paradigm shift is that more and more journals want to see it before they publish a genetic study.
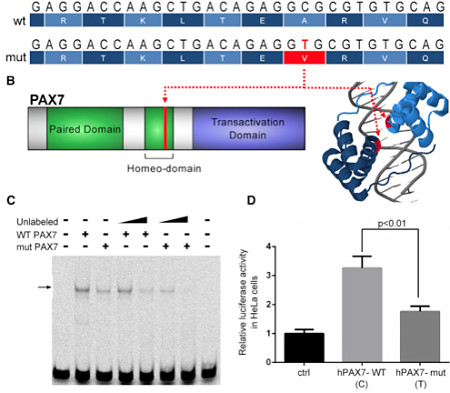
PAX7 de novo Mutation
One of the few de novo coding mutations was predicted to disrupt the DNA-binding domain of PAX7. The authors designed an electromobility shift assay (EMSA) to examine how the missense substitution affected PAX7‘s ability to bind a target regulatory sequence. They also used quantitative reporter assays in HeLa cells, with co-transfection of a plasmid containing either wild-type or mutant PAX7.
PAX7 functional validation (Leslie et al, AJHG 2015)
Both experiments showed that the wild-type allele had greater DNA-binding capacity, and drove higher expression of the reporter gene.
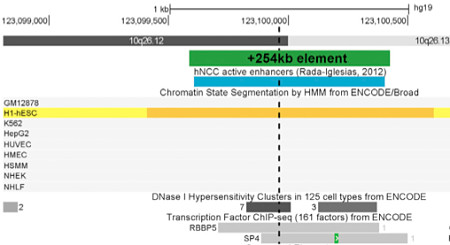
FGFR2 de novo Mutation
One of the noncoding de novo mutations was 254 kilobases downstream of FGFR2, in a noncoding region that looks (according to chromatin marks) like a neural crest enhancer. FGFR2 is known to play a role in craniofacial development, and rare variants in it had been reported in cases of NSCL/P. Here, the authors leveraged a zebrafish model system to examine the role of that enhancer during development. In transient transgenic reporter studies of zebrafish embryos, the +254kb element holds up: the wild-type allele had enhancer activity in 41/82 embryos (50%), whereas the mutant allele had enhancer activity in 3/83 embryos (3.6%).
FGFR +254kb enhancer (Leslie et al, AJHG 2015)
This, in my opinion, is one of the most compelling parts of this study: in vivo functional validation of a single base change in a noncoding enhancer that’s hundreds of thousands of bases away from the gene it regulates.
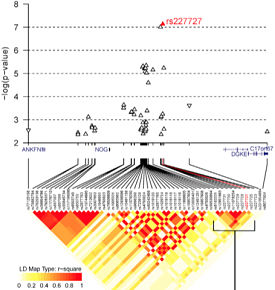
Common Variant at 17q22 (NOG)
Multiple SNPs reached genome-wide significance in the 17q22 region. The greatest significance was detected at rs227727, about 105 kb downstream of the NOG transcriptional start site. This variant was in complete LD with the lead SNP from the prior GWAS. This was interesting because NOG encodes a BMP antagonist that’s expressed primarily in the epithelium during palatal development.
Tandem enhancer disruption (Leslie et al, AJHG 2015)
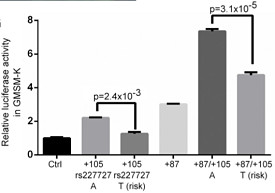
The authors confirmed NOG expression in the palatal epithelium in mouse embryos. They also noted that rs227727 mapped to one of two enhancers in the region, +105kb (the other being +87 kb). The variant allele disrupts predicted binding sites for two transcription factors (MEF2C and CDX2) and creates possible binding sites for at least two others.
Interestingly, the zebrafish assay did not show epithelial enhancer activity for the +105kb element by itself. However, a tandem construct with both enhancers (+87kb and +105kb) lit things up. The effect was at least additive, and constructs containing the risk allele of rs227727 showed significantly decreased enhancer activity.
Beyond GWAS for Complex Disease
What I like about this study is that it studied GWAS and candidate gene regions in careful investigations that included functional validation components. It’s so easy to take a GWAS hit, look for the nearest gene, and spin a story about how variation in that gene affects the phenotype of interest. Here, the authors have done the difficult and time-consuming work of (1) exhaustive sequencing to identify the possible functional variants, and (2) in vivo functional assays to prove that the implicated variants have a phenotypic effect. That’s a lot of work to pin down the genetic architecture and disease mechanism for a handful of disease loci.
The high-throughput nature of genotyping (and increasingly, sequencing) and the discovery power of large cohorts are going to yield promising new findings. With them comes a strong temptation to take the association hits and run with them. Write up some voodoo in the discussion about the gene and its role, get the paper out, and move on. The problem is that statistical genetic evidence is not enough. You don’t know that your lead SNP is functional, or that the nearest neighboring gene provides the mechanism of phenotypic effect.
More studies like these, with well-planned study designs and compelling functional assays, will be required as we continue to unravel the complex fabric of human genetics.
References
Leslie, E., Taub, M., Liu, H., Steinberg, K., Koboldt, D., Zhang, Q., Carlson, J., Hetmanski, J., Wang, H., Larson, D., Fulton, R., Kousa, Y., Fakhouri, W., Naji, A., Ruczinski, I., Begum, F., Parker, M., Busch, T., Standley, J., Rigdon, J., Hecht, J., Scott, A., Wehby, G., Christensen, K., Czeizel, A., Deleyiannis, F., Schutte, B., Wilson, R., Cornell, R., Lidral, A., Weinstock, G., Beaty, T., Marazita, M., & Murray, J. (2015). Identification of Functional Variants for Cleft Lip with or without Cleft Palate in or near PAX7, FGFR2, and NOG by Targeted Sequencing of GWAS Loci The American Journal of Human Genetics DOI: 10.1016/j.ajhg.2015.01.004