Few areas of biomedical research have benefited more from next-gen sequencing than studies of rare inherited diseases. Rapid, inexpensive exome sequencing in individuals with rare, presumably-monogenic diseases has been hugely successful over the past few years. There’s been a lot of discussion in the NGS community about the analysis burden of the large-scale whole-genome sequencing that will be possible with Illumina HiSeqX Ten systems, but even exome sequencing analysis brings considerable challenges.
In the March 2014 issue of the American Journal of Human Genetics, we present a software package called MendelScan to aid the analysis of exome data in rare Mendelian disorders.
Exome Sequencing Challenges
Every individual harbors thousands of coding variants, 5-10% of which are not in public databases such as dbSNP. A study led by my friend Daniel MacArthur found that, even after correcting for annotation errors and other artifacts, the genome of a healthy individual contains ~100 loss of function coding variants. This is just one of the reasons that exome sequencing of Mendelian disorders can fail.
And they do fail. The current solve rate for incoming cases at NIH Mendelian Centers remains at around 25%. Dominant disease pedigrees remain more difficult to solve than recessive ones.
Mendelian Disorder: Retinitis Pigmentosa
Retinitis pigmentosa (RP) offers a wonderful example of challenging Mendelian disorders. It’s a “genetically heterogeneous” disease, which is a fancy way of saying that many different mutations in dozens of different genes can cause dominant, recessive, or X-linked disease. The disease affects around 1 in 3,500 individuals in the U.S., and it’s incurable.
No matter the genetic cause, the progression of RP is remarkably uniform. Basically, it’s a disease of rod photoreceptors — the light sensing kind, not the color-sensing kind — whose slow, inexorable attrition usually causes night blindness (usually apparent by adolescence) and a sustained narrowing of the visual field (tunnel vision). Most RP patients will be legally blind by the age of 40.
Mutations in about 18 different genes can cause dominant RP, which is the form that we’re studying. Routine genetic testing of common disease-causing mutations explains about 50% of incoming cases right off the bat. We’re interested in the cases that come back negative from these screens, the ones that may have rare or as-yet-unknown causal mutations.
Exome Sequencing in 24 Families
We did exome sequencing for 24 families that lacked common disease-causing mutations. The typical family had a proband, the affected parent (because it’s dominant), the unaffected parent, and a distant affected relative. Overall we did 2-7 affected and 0-2 unaffected samples per family, for a total of 91 samples. On average, in each family, we identified ~30,000 single nucleotide variants (SNVs) and 600 insertions/deletions (indels) in coding regions. That’s a lot to sort through when you’ve got 24 families.
Variant Prioritization Strategy
Based on our knowledge of dominant RP, and an analysis of 762 disease-causing mutations downloaded from HGMD, we expected that most disease-causing mutations would exhibit some key characteristics:
- Segregation. In dominant pedigrees with full penetrance, all affected individuals should carry the causal mutation, and none of the unaffected individuals should.
- Rareness. All of the mutations known to cause dominant RP are quite rare. In the HGMD set, 68% of mutations were novel to dbSNP 137, and another 21% were present only because they were pulled in from OMIM and other mutation databases.
- Protein impact. We expect that most (but not all) causal mutations will impact genes. When classified by current VEP annotation, most of the mutations were predicted to alter protein sequence (66%), reading frame (13.5%), splicing (4.3%), or length (6.8%).
- Retinal expression. Genes in which mutations cause retinal disease tend to be highly expressed in the retina. According to recent human retina RNA-seq data, about 97% of genes in RetNet (a retinal disease gene database) are in the top 50% of all genes when ranked by retinal expression.
You’ll note that there are exceptions to every rule above. That’s why we were uncomfortable with simply ruling out variants that don’t segregate perfectly or ones that appear to be synonymous. Instead, we developed a scoring algorithm to prioritize variants based on segregation, rareness, annotation, and retinal expression.
So how well does it work? In our exome dataset, 8 of 24 families harbored a likely-pathogenic mutation in a known RP gene. When we sorted the variants in those families by prioritization score, the causal mutation never ranked lower than 13. Out of 20,000+ variants. There was one exception, a family that turned out to have an error in the pedigree. The causal mutation there ranked #439 out of 26,666 SNVs, so it was still in the top 2%.
This robust performance — even in the face of incorrect assumptions — is why we prefer to prioritize rather than filter-and-remove candidate variants.
Mapping Dominant Disease Genes
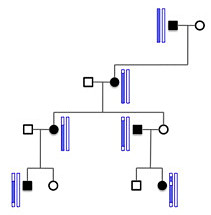
Koboldt et al, AJHG 2014
Even though our scoring algorithm seemed to be working well, we still had hundreds or thousands of variants to sift through in some families. And while those pedigrees often weren’t large enough for traditional linkage analysis, we asked whether the dense information provided by exome sequencing could help nominate or exclude regions based on segregation.
Disease-causing variants usually don’t occur in isolation. They’re part of a haplotype that segregates within a family pedigree (example at right). For dominant disease, all affected individuals have one haplotype in common, the one that hosts the causal variant (denoted in black, on the left):
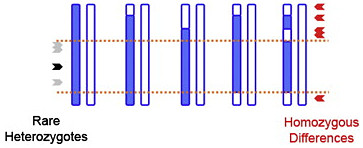
Rare Heterozygote Rule Out
That haplotype (orange) might also host other variants that aren’t disease-causing, but might still be picked up by exome sequencing. Some may be quite rare, and because they’re physically linked to the causal mutation, they’ll be heterozygous in affected individuals. There also should be no homozygous differences between pairs of affecteds (red) because all affecteds share at least one haplotype. So a cluster of shared rare (heterozygous) variants, and an absence of homozygous differences, helps us map haplotypes shared by affecteds in the pedigree. We call this rare heterozygote rule out (RHRO).
Shared IBD Analysis
Another approach would take the same principles, but use identity by descent (IBD). Since the haplotype shared by affecteds was inherited from a common ancestor, we can also search for regions that are IBD between most or all pairs of affecteds. We call this shared IBD (SIBD) analysis, and like RHRO, its discriminatory power grows with the numbers of and genetic distance between affecteds in a pedigree.
We applied these approaches to families with 3+ sequenced affecteds. When you put both mapping methods together, you get something like this:
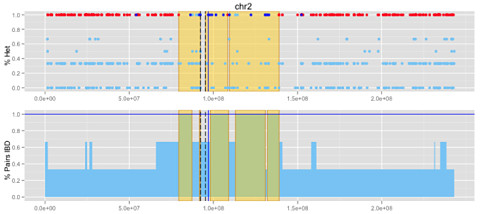
Koboldt et al, AJHG 2014
Some of them had orthogonal information — traditional linkage peaks or an identified pathogenic mutation — that told us where the disease-causing variant resided (blue line, above), so we could test the performance of these approaches. Our mapping methods did well: they recapitulated known linkage regions and/or captured the region of the causal mutation. And they did so with far fewer affected individuals than were used for the linkage analysis.
Our approaches also identified new candidate regions. These might be eliminated by adding more affected individuals, or they might reflection linkage that was missed by traditional approaches.
Improving Exome Analysis for Rare Disorders
So the MendelScan tool is out and freely available. We would love to have your feedback and suggestions for it! The current JAR release is v1.2.1. Let’s go forth and conquer some Mendelian disorders.
References
Koboldt DC, Larson DE, Sullivan LS, Bowne SJ, Steinberg KM, Churchill JD, Buhr AC, Nutter N, Pierce EA, Blanton SH, Weinstock GM, Wilson RK, & Daiger SP (2014). Exome-Based Mapping and Variant Prioritization for Inherited Mendelian Disorders. American journal of human genetics PMID: 24560519