
Stergachis et al (Science 2013)
A recent paper in Science has been hyped as the revelation of a new code affecting codon choice and protein evolution. In their study, Andrew Stergachis et al from the University of Washington applied DNAaseI-seq to map transcription factor occupancy across the human exome in 81 different cell types. They found that around 15% of codons direct TF binding in addition to protein sequence.
These dual-use codons, which the authors give the [unfortunate] name “duons”, appear to drive codon usage bias and are under evolutionary constraint. A signification fraction of SNVs within duon affect the transcription factor binding, suggesting yet another way that even synonymous coding variants may affect a molecular phenotype. In this post I’ll dig into the study and discuss what it adds to our understanding of genome content and function.
Genome-wide DNaseI Footprinting
The authors first undertook a genome-wide approach, called DNAaseI footprinting, to identify regions of protein-DNA interaction in 81 diverse cell types. This technology combines the digestion of DNA with a restriction enzyme (DNaseI) with next-gen sequencing. Regions of the genome that inactive (“closed” chromatin, also called heterochromatin) are tightly packaged around nucleosomes and histone proteins, which protects them from cleavage. Active genomic regions, in contrast, are open (euchromatin) and thus systematically cleaved by DNAaseI.
Stergachis et al (Science 2013)
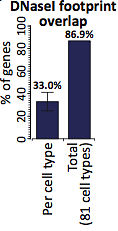
What DNAaseI footprinting ultimately yields is a series of broad peaks of DNaseI sensitivity (high depth of DNaase-Seq reads) surrounding smaller “footprints” (low depth of DNase-Seq reads) where the transcription factors are actually bound. Collectively, the authors identified about 11.6 million footprints genome-wide, ranging in size from 6-40 bp.
Interestingly, 216,304 of those footprints were completely within a protein-coding exon. That’s only about 1.8% of the footprints, but it hinted at gene-regulatory sequences that overlap coding sequence. About 14% of all human coding bases bound a TF in at least one cell type, and these interactions affected the vast majority (87%) of protein-coding genes.
Targeted Exonic Footprinting
Stergachis et al (Science 2013)
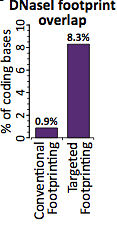
To pursue more comprehensive DNaseI footprinting of coding sequences, the authors developed a targeted approach using RNA probes complementary to human exons, i.e. Agilent exome-like capture of DNase-Seq libraries. This yielded about a 10-fold increase in exonic DNaseI cleavage, reaching a sensitivity that would have required about 4 billion DNase-Seq reads if done genome-wide.
Overall, there were about 175,000 coding DNaseI footprints per cell type. Given this abundance, the authors wanted to know if the footprints were distributed in a particular way, and if they were under evolutionary constraint (i.e. being acted upon by natural selection).
Distribution of Coding DNaseI Footprints
So, the results indicated that some regulatory proteins — presumably transcription factors — were binding DNA in coding regions. This is not a novel finding; the ENCODE group made a similar observation some time ago using DNAase I and CHiP-Seq data.
Stergachis et al (Science 2013)
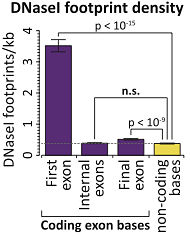
However, the authors of this current study dug deeper. They examined the density of footprints across exons, genes, and the rest of the genome. Internal exons were not significantly different from noncoding bases in terms of footprint density, but the first and last exons were.
The most striking pattern was a high density of DNAaseI footprints in the first coding exon. Otherwise, the density varied widely and the actual number of footprints per gene was a function of gene’s size and expression level.
Evolutionary Constraint of Coding Footprints
Next up, analyses of how conserved coding DNaseI footprints are to the rest of the exome and gene. Here, the authors looked at four-fold degenerate bases (4FDBs), which are those third bases in codons that can change without altering protein sequence. Presumably, such bases are under neutral evolution, since a substitution won’t alter the protein. At least, in theory. There are considerations like tRNA abundance that may come into play.
Stergachis et al (Science 2013)
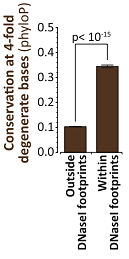
As a proxy for conservation, the authors used mean the mean phyloP value. This value is between 0 and 1, representing the extent to which a base is conserved (calculated by PHAST) across vertebrate genomes. I assume vertebrate genome alignments are the source, though the authors don’t explicitly state where they got the phyloP values (come on, peer reviewers!). Most likely, they downloaded phyloP values from the UCSC Genome Browser database.
In any case, phyloP values were significantly higher (on average) for 4FDBs within DNaseI footprints than outside of them, suggesting a higher signal of evolutionary constraint. The authors argue that, since selection would not be acting on amino acid changes at 4FDBs, it must be acting on the sequence-specific binding of transcription factors instead.
Mutation Age and Conservation
As another way of looking at evolutionary constraint, the authors examined the estimated age of coding mutations using data from the Exome Sequencing Project (4300 European-origin and 2200 African-origin individuals). The underlying principle here is that mutations in regions under selection will have occurred more recently than those in neutrally-evolving regions. From Figure 1:

Age of Coding SNVs (Stergachis et al, Science 2013)
In general, coding SNVs that were inside DNaseI footprints were younger than those outside footprints, lending further support to the idea that these regions are evolutionarily conserved. Interestingly, this observation holds true regardless of whether the mutation was synonymous (black) or nonsynonymous (red). We already know that nonsynonymous mutations are under selection, so this evidence would suggest that both TF binding and amino acid coding are “constrained” by evolution in coding regions.
Codon Bias in TF Binding
Stergachis et al, Science 2013
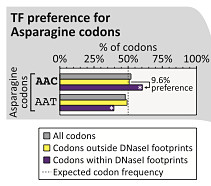
Most amino acids are encoded by two or more triple-base combinations, though it’s been known for some time that there’s sometimes a preference for one codon over the others when you look at the actual coding sequences of known genes. For example, 52% of asparagine residues in human proteins are encoded by AAC, whereas 48% are encoded by AAT.
Interestingly, the authors observed that TF binding (inferred from DNaseI footprints) seems to reinforce the genome-wide preference for certain codons. In Asparagine codons overlapped by such footprints, for example, the preference for AAC rose to around 60%. This held true for most amino acids encoded by two or more codons. In other words, the codon preferred for protein-coding was also preferentially bound by transcription factors. The authors argue that TF binding is a major driver of this bias.
Overall, this makes for an intriguing study as it highlights the importance of TF binding in protein-coding regions. Conservation and codon usage analyses suggest that such bindings are not happy accidents, but actually drivers of codon preference and a source of additional phenotypic variation. Is it a third code? Not really, because transcription factors (for the most part) vary in which motifs they’ll bind, and when. In contrast, triplet codons always specify a certain amino acid. Still, it’s a way that even synonymous variation in coding regions may induce a molecular phenotype, and thus these “silent” variants should merit further consideration.
References
Stergachis AB, Haugen E, Shafer A, Fu W, Vernot B, Reynolds A, Raubitschek A, Ziegler S, LeProust EM, Akey JM, & Stamatoyannopoulos JA (2013). Exonic transcription factor binding directs codon choice and affects protein evolution. Science (New York, N.Y.), 342 (6164), 1367-72 PMID: 24337295