It is increasingly clear that the annotation and interpretation of sequence variants represents one of the most important challenges in human genetics. With next-gen sequencing, our ability to identify variants — in disease pedigrees, case-control cohorts, and apparently healthy individuals — has rapidly outpaced our ability to say anything about what those variants do. This is a problem that must be tackled from several different angles, notably:
- Genetic studies. Common variant association studies (CVAS), sequencing-enabled rare variant association studies (RVAS), and family studies identify variants associated with a phenotype, such as disease susceptibility.
- Genome characterization. The ENCODE Project and similar efforts characterize elements of the genome (genes, regulatory sequences) that may have functional relevance.
- Functional studies. Measuring cellular/tissue phenotypes, such as gene expression, and correlating them to genetic variation helps elucidate the molecular mechanisms connecting genotype to phenotype.
An article just published in Nature exemplifies the kind of powerful functional studies enabled by large-scale, high-throughput sequencing efforts. Tuuli Lappalainen and colleagues performed mRNA and microRNA sequencing in 462 lymphoblastoid cell lines (LCLs) from the 1,000 Genomes cohort. The individuals come from five populations, four of European origin (CEPH Europeans, Finns, British, and Toscani Italians) and one of African origin (Yoruba Nigerians). Most of these samples were sequenced during the first phase of the 1,000 Genomes Project; the remainder were imputed using SNP array data.
Lappalainen et al Nature 2013
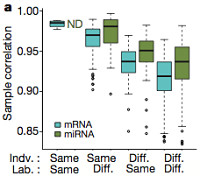
RNA Sequencing: Mature Technology
Seven different labs performed the RNA sequencing for this study. A comparison of data for 5 individuals sequenced in replicate (in different labs) demonstrated that variation between individuals was greater than variation between labs. The authors therefore feel that the technology has matured enough to allow distributed RNA-Seq production. This does not mean there will be no batch effects, of course. There are always batch effects with next-gen sequencing, even within centers.
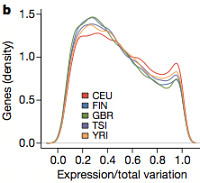
Transcriptome Variation
Lappalainen et al Nature 2013 (Fig 1b)
This study offered a nice dataset to look at gene expression variation within and between populations. They looked at two categories of differences between individuals and groups:
- The overall gene expression level
- The relative abundance of different transcripts for the same gene (transcript ratios)
The relative contribution of these seems to be characteristic for each gene, and similar across populations, with the second category (transcript ratio) contributing more to total variation on average.
Population differences explained only about 3% of the variation; in European-European comparisons, transcript ratios account for 6-40% of differences, but between Africans and Europeans, 75-85% of differences were in transcript ratio. This hasn’t been seen before in humans, although previous studies in mammals and vertebrates have shown that splicing patterns better distinguish different species than overall gene expression levels.
Expression Quantitative Trait Loci
The authors performed a deep analysis of expression data (mRNA/miRNA) with genotypic data (SNPs/indels) to identify variants influencing gene expression, commonly called expression quantitative trait loci (eQTLs). Classical eQTLs were observed for 3,773 protein-coding or noncoding long RNA genes. These are genes for which variants are associated with overall expression. Going deeper, and looking at individual exons, more than doubled the number of eQTLs to 7,825. In this set, when the authors regressed out the first eQTL, around a third of genes had a second, independent eQTL and it was usually associated with a different exon.
To look specifically at genetic effects on splicing, the authors sought transcript ratio QTLs (trQTLs) in which variants were correlated to the ratio of different transcript usage, finding 639. There were 279 genes with both types (eQTL and trQTL); regression analysis showed that for 57% of these, the correlated variants were independent of one another. This suggests that the regulatory variants are largely different.
Analyzing microRNAs and mRNAs
It could be my own biased view of the world, but it seems that interest in microRNAs (miRNAs) has waned slightly in the NGS community. They’re obviously important but notoriously difficult to study, and especially to implicate in human disease. And the number of reported human miRNAs in MiRbase is now over 1800. In this study, however, only 644 autosomal microRNAs were quantified (in >50% of individuals) by small RNA sequencing. That seems to be around the magic number.
Because miRNAs supposedly regulate gene expression, the authors performed mRNA-miRNA analysis in a steady-state sample to identify likely interactions. Of 100 miRNA “families”, 32 correlated with the expression of predicted target exons. Strangely, 45% of those correlations were positive, despite the general belief that miRNAs downregulate target genes. When the variants correlated with microRNA or mRNA expression were examined — by examining if SNPs associated with the expression of a nearby miRNA were also associated with that miRNA’s predicted target — the only significant results were found when the canonical, negative correlation pattern (more microRNA = less mRNA) was observed.
The authors note that non-canonical, positive correlations were over-represented in mRNAs for transcription factors. So the positive correlations might results from a kind of feedback loop in which miRNAs and their targets are up-regulated together under steady-state conditions.
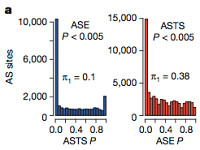
Characterizing Regulatory Variation
Lappalainen et al, Nature 2013
Perhaps the strongest aspect of this study is how the authors leverage their expression-and-genotype dataset to look at regulatory variants in the genome. They identified two types of allelic effects:
- Allele-specific expression (ASE), observed in 6.5% of sites per individual
- Allele-specific transcript structure (ASTS), observed in 5.6% of sites per individual.
There was a substantial amount of overlap, which could mean that transcript structure differences drive the variation in gene expression levels. Notably, the vast majority of ASE/ASTS sites were very rare in populations. This suggests that a substantial portion of gene expression regulation is achieved by rare variants that won’t be detected by eQTL analysis, both because of insufficient power and because rare variants aren’t represented on SNP arrays.
Genetic or Epigenetic Regulation?
Next, the authors sought to address the question of whether or not allele-specific expression patterns were due to epigenetic patterns. Their approach is clever:
- Identify transcripts exhibiting biased expression favoring one allele (allele-specific expression, or ASE)
- Look for nearby variants associated with that transcript’s expression
- Identify the subset of those in which ASE is observed in individuals heterozygous for the variant, but not observed in individuals that are homozygous. If the variant’s homozygous but ASE is evident, they’re independent of each other.
Overall, about 7.4% of the quarter million variants tested seem to be putative regulatory SNPs. Among 5,479 variants exhibiting allele-specific expression; for 95% o them, there was an enrichment of putative regulatory SNPs within 100 kbp. This suggests that a large proportion of allele-specific expression has a genetic (i.e. not epigenetic) basis. Undoubted, haplotype-specific epigenetic patterns have some effect on gene expression, but it’s less than that of the sequence variants themselves.
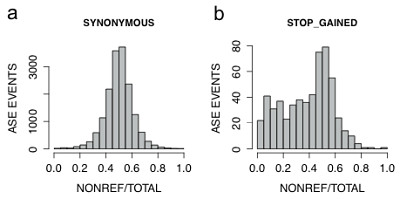
Loss of Function Effects
As a research community, we rely heavily on computationally-driven annotation of genetic variants to infer their likely pathogenicity. This dataset offers the opportunity to evaluate the true impact of such predictions on the molecular phenotype of gene expression. There were 839 nonsense and 849 splice-site variants captured in the RNA sequencing dataset. As expected, nonsense variants tend to induce nonsense-mediated mRNA decay (NMD):
Lappalainen et al, Nature 2013
Also as expected, nonsense variants near the end of the transcript are less likely to trigger decay. Intriguingly, however, 68% of variants predicted to trigger nonsense-mediated decay show equal representation of both alleles in the RNA sequencing data. So there must be other, as-yet-unknown mechanisms of escaping NMD.
For the splice variants, the effects observed in RNA-Seq data were closer to what’s expected. About 10% of the splice variants are predicted to destroy the splice site motif, and individuals homozygous for those exhibited a ~29% lower inclusion rate of the affected exon. There’s still a lot going on with splicing that remains to be characterized, but at the very list, this confirms that the very rare, obviously disruptive splice site variants have a significant impact on transcript structure.
Summary
Overall, this integrated analysis offers a good model for helping connect sequence-level genotype to molecular phenotype. The findings are informative, and suggest a lot of promise for regulatory variation underlying important traits. More studies like these are needed, especially in disease cohorts.
Be sure to also read the guest post by lead author Tuuli Lappalainen over at Genomes Unzipped for more insight into this work.
References
Lappalainen T, Sammeth M, Friedländer MR, ‘t Hoen PA, Monlong J, Rivas MA, Gonzàlez-Porta M, Kurbatova N, Griebel T, Ferreira PG, Barann M, Wieland T, Greger L, van Iterson M, Almlöf J, Ribeca P, Pulyakhina I, Esser D, Giger T, Tikhonov A, Sultan M, Bertier G, MacArthur DG, Lek M, Lizano E, Buermans HP, Padioleau I, Schwarzmayr T, Karlberg O, Ongen H, Kilpinen H, Beltran S, Gut M, Kahlem K, Amstislavskiy V, Stegle O, Pirinen M, Montgomery SB, Donnelly P, McCarthy MI, Flicek P, Strom TM, Geuvadis Consortium, Lehrach H, Schreiber S, Sudbrak R, Carracedo A, Antonarakis SE, Häsler R, Syvänen AC, van Ommen GJ, Brazma A, Meitinger T, Rosenstiel P, Guigó R, Gut IG, Estivill X, Dermitzakis ET, Palotie A, Deleuze JF, Gyllensten U, Brunner H, Veltman J, Cambon-Thomsen A, Mangion J, Bentley D, & Hamosh A (2013). Transcriptome and genome sequencing uncovers functional variation in humans. Nature, 501 (7468), 506-11 PMID: 24037378