
Image Credit: Nature (1000 Genomes)
In the ten years since the first draft of the human genome was published, our knowledge of the relationship between genotype and phenotype has grown exponentially. Perhaps the most important achievement of the genome assembly is that it provided a reference, against which individual DNA sequences could be compared to identify sequence variation.
The sequel to the Human Genome Project, the International HapMap Project, set out to build a map — specifically, a haplotype map — of common genetic variation in several world populations. And the sequel to that project, the 1,000 Genomes Project, sought to characterize the nature and extent of genetic variation, especially variants that were rare (MAF<0.05).
HapMap and Genome-Wide Association Studies
The completion of the HapMap marked an important turning point for human genetics: the possibility of genome-wide association studies (GWAS). With this map, and the high-throughput genotyping technologies that developed during the HapMap, it became feasible to rapidly genotype many samples at thousands of polymorphic, informative positions throughout the genome. And because these markers were chosen on the basis of linkage disequilibrium information, they managed to capture (represent) much of the variation in each individual.
In the years that followed, researchers used GWAS to study a multitude of diseases and phenotypes. Many new genetic associations arose, as illustrated by this figure from the NHGRI GWAS database.
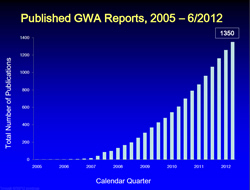
GWAS reports from 2005-2012 (Credit: NHGRI)
At the same time, it became apparent that common variants alone did not explain the genetic component of most complex human diseases. And from the 1,000 Genomes Project, we began to understand that a significant proportion of variants in any individual genome (5-10%) have never been seen before.
Rise of Rare Variants
It has become clear that these rare variants are equally important for understanding the genetic basis of human disease. Next-generation sequencing makes it possible to identify these in individual samples, but understanding their contribution to phenotype presents some challenges:
- By definition, rare variants will only be found in a small fraction of sequenced samples.
- Large cohorts will be required to assess genetic association of rare variants with a phenotype. We’re talking about thousands or tens of thousands of individuals.
- Sequencing large numbers of samples, especially whole-genome sequencing, remains costly. Yes, I know, NGS is so cheap now. But when you multiply the cost for WGS (~$3,000) by the number of samples (say 1,000), suddenly we’re talking about $3 million.
Currently, researchers are employing a number of strategies to get results without spending millions of dollars. In general, these strategies fall into two categories:
- Targeted sequencing, where only certain regions of the genome are sequenced (i.e. the exome)
- Fewer samples, with the idea of using statistical methods to address insufficient power
Special Frontiers Issue: Rare Variants in Human Disease
The development of statistical and analytical methods to uncover rare variants associated with human disease is a rapidly evolving field. This summer, I am helping edit an issue of Frontiers (now Nature Frontiers) entitled Identification of rare genetic variants contributing to human diseases. The deadline for abstracts is July 1. Desired topics include:
- Calling, genotyping, QC, and validation of rare variants
- Discovery of novel variants and de novo mutations
- Annotating and predicting function of rare variants
- Methods for discovering rare causal variants in Mendelian disease.
- Methods for associating rare variants with complex disease
- Burden and non-burden test methods for genes, pathways and other self-defined variant sets;
- Estimating rare variant effect size and heritability
- Integrative analysis of rare and common variants;
- Applying rare variant analysis to specific diseases
For details, please visit the special issue’s page on the Frontiers web site.